


INTRODUCTION
Systemic sclerosis (SSc) is a chronic autoimmune disorder characterized by endothelial and fibroblast dysfunction, resulting in progressive fibrosis of the skin and internal organs, more frequently, the lungs and gastrointestinal tract [1]. Although the pathogenesis of SSc is not well understood, it is considered an autoimmune disorder due to the presence of autoantibodies. In contrast, ankylosing spondylitis (AS) is considered an autoinflammatory disease, as it involves the innate immune system [2]. Although the prevalence rates of AS and SSc were reported as 12.2-18.6 per 10,000 patients [3] and 31-286 per 1,000,000 patients [4], respectively, an association between SSc and AS is rarely observed, as the two diseases share few characteristics.
Patients with SSc frequently have arthritis in the peripheral joints. Interestingly, sacroiliac joint involvement is also found in about 22% of SSc patients [5]. However, in some cases there are no joint symptoms of SSc, but there are manifestations of AS [6-10]. Although the two diseases manifested independently in our patient, there seem to be common features among patients with both diseases. Analysis of these cases may help to elucidate the association between the two diseases. We present a case of SSc in a 44-year-old man who also had a diagnosis of AS, and present a review of similar cases in the literature.
CASE REPORT
A 44-year-old man was referred to the Department of Rheumatology for the known AS. The patient had previously been diagnosed with AS based on symptoms of chronic inflammatory back pain, human leukocyte antigen (HLA)-B27 positivity, and pelvic X-ray findings. The patient had been followed by a rheumatologist for 18 years. At the time of the visit, the patient had no inflammatory back pain or peripheral arthritis. He also had no enthesitis, uveitis, psoriasis, or inflammatory bowel disease associated with AS. HLA-B27 test was positive, and grade 2 bilateral sacroiliitis was observed on pelvic radiographs (Fig. 1A). The patient was diagnosed with AS according to the Assessment of SpondyloArthritis international Society criteria. [11]. The disease activity of AS was 2.5, as assessed using the bath ankylosing spondylitis disease activity index.
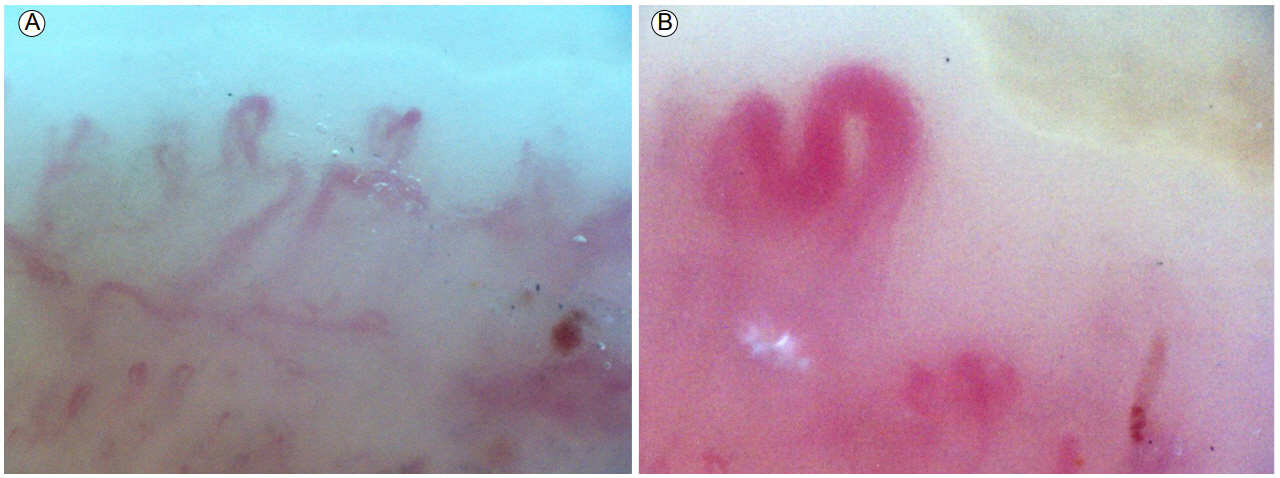
The patient visited the rheumatology clinic 5 months ago for Raynaud phenomenon and severe coughing. He had periungual telangiectasia, but there were no pitting scars. Neither puffy changes nor digital ulcers were observed in the fingers. There was no telangiectasia or calcinosis of the skin. The skin of the face (modified Rodnan skin score 1), both hands (2), and fingers (2) showed mild thickening, and the total modified Rodnan skin score was 5. Late scleroderma pattern characterized by loss of capillaries, and ramified (Fig. 2A) and enlarged (Fig. 2B) capillaries, was observed by nailfold capillaroscopy. The patient was diagnosed with SSc according to the 2013 classification criteria [12]. Based on clinical features of skin involvement, the patientŌĆÖs SSc was thought to be of the limited pattern type.
Laboratory examination revealed white blood cell count of 5,000/╬╝L (range: 4,000-10,000/╬╝L), hemoglobin level of 13.5 g/dL (13.0-17.0), and platelet count of 205,000/╬╝L (150,000-450,000/╬╝L). Alanine transferase and aspartate aminotransferase levels were 20 IU/L (5-40) and 22 IU/L (5-45), respectively. The albumin level was 4.2 g/dL (3.2-5.5) and the total protein concentration was 7.7 g/dL (6.4-8.5). The erythrocyte sedimentation rate was 15 mm/h (0-15) and the C-reactive protein level was < 0.8 mg/dL (0-0.8). Immunological tests for differential diagnosis of SSc were performed. The anti-nuclear antibody level was 1:2,560 with a dense, fine-speckled pattern. Anti-Ro antibody and anti-Scl-70 antibody tests were positive, but anti-La antibody and anti-centromere antibody tests were negative. Chest computed tomography indicated interstitial lung disease (ILD) with a nonspecific interstitial pneumonia pattern and pleural effusion in the right lung (Fig. 1B). There was a noncalcified nodule in the lateral basal segment of the left lower lobe, and multiple calcified and noncalcified lymph nodes were seen in the mediastinum and retroperitoneum. Spirometry showed a mild restrictive pattern with forced expired volume in 1 second/forced vital capacity (FEV1/FVC) of 94.93%, FVC of 68.6%, and FEV1 of 77.2%. Carbon monoxide diffusion capacity was 48.1%. The patient was treated with diltiazem (90 mg) and aspirin (100 mg) for management of Raynaud phenomenon. However, no specific management was applied for the minimal ILD.
Three months later, the patientŌĆÖs cough became worse. Loculated pleural effusion that appeared in the right lower lung was observed on chest X-ray. Percutaneous catheter drainage was performed for differential diagnosis of the pleural fluid. A total of 250 mL of bloody fluid was drained over 5 days. Tests of the pleural fluid showed protein level of 5 g/dL, albumin level of 2.6 g/dL, lactate dehydrogenase level of 639 U/L, and glucose level of 100 mg/dL. Pleural fluid analysis revealed exudative fluid with neutrophil dominance (72%) and no microbial organisms. There was no evidence of tuberculosis on either the acid-fast bacilli test or polymerase chain reaction. These findings suggested that the pleural effusion was caused by inflammation rather than infection. After drainage of the pleural fluid, the patientŌĆÖs cough subsided, and he was discharged with improved symptoms.
DISCUSSION
There have been several reports regarding the combined occurrence of SSc and AS (Table 1) [6-10]. Most of the clinical characteristics in other cases were similar to those in our patient, with the exception of limited skin involvement and low-grade sacroiliitis without peripheral arthritis. Although SSc generally shows female predominance and AS shows male predominance [3,4], it is interesting that all reported cases of simultaneous SSc and AS were male. Male predominance was also apparent in a study of the presence of sacroiliitis in patients with SSc [5].
Common findings of skin thickening, ILD, and anti-nuclear antibody positivity were observed in patients with SSc. All patients also showed specific features of AS, including sacroiliac joint involvement and HLA-B27 positivity. As the onset of AS symptoms generally begins at a young age, it seems natural to assume that AS develops earlier than SSc or other connective tissue diseases. Thus, SSc appears to be associated with AS in middle-aged male patients. In addition, ankylosis was progressive and associated with diffuse SSc with ILD. However, in our case, AS was not severe, and SSc was present in a limited pattern.
The association between AS and connective tissue diseases could be attributable to linkage disequilibrium between the HLA-B27 and HLA DR4 antigens [7,13]. Kayser et al. [8] demonstrated an association of SSc and AS with HLA, and suggested a ŌĆ£genetic trapŌĆØ in which distinct genes may act together, resulting in increased susceptibility to two different HLA-associated systemic diseases. Considering this suggestion, it is interesting that patients with calcinosis, RaynaudŌĆÖs phenomenon, esophageal dysmotility, sclerodactyly, and telangiectasia (CREST) syndrome have a higher frequency of HLA-B27 [14].
Thompson studied the prevalence of pleural effusion in SSc patients. Pleural effusions were identified in only 7% of patients, which was a very low prevalence in comparison to that of systemic lupus erythematosus [15]. Pleural effusion was found to often accumulate in patients with pulmonary artery hypertension associated with SSc and right-sided heart failure [16]. Pleural effusion in AS also occurs very rarely. Rosenow et al. [17] reported three cases (0.15%) after reviewing the chest X-rays and reports of 2,080 AS patients. Although inflammative pleural exudate was observed in our patient, it was difficult to determine the cause.
In conclusion, we reported a very rare case of combined SSc and AS. Manifestations of SSc, including skin thickening, Raynaud phenomenon, and ILD, were observed along with the features of AS. In addition, pleural effusion, which is rare in both SSc and AS, was present in the case. Accumulation of further case series data will be needed to determine the common etiology of the two diseases.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






