


ņä£ ļĪĀ
Ļ░äņ¦łņä▒ ĒÅÉņ¦łĒÖś(interstitial lung disease, ILD)ņØĆ ĒÅÉņŗżņ¦łņØä ņ╣©ļ▓öĒĢśļŖö ļ╣äĻ░ÉņŚ╝ņä▒, ļ╣äņóģņ¢æņä▒ ņ¦łĒÖśņ£╝ļĪ£ņä£ ĒÅÉĒż ņé¼ņØ┤ Ļ│ĄĻ░äņØĖ Ļ░äņ¦ł(interstitium)ņŚÉ ņäĖĒżņØś ņ”ØņŗØĻ│╝ ļČäĒÖö, ļ¦īņä▒ņĀüņØĖ ņŚ╝ņ”Ø ļ░Å ņä¼ņ£ĀĒÖö ļō▒ ļŗżņ¢æĒĢ£ ļ│æļ”¼ ĻĖ░ņĀäņØ┤ ļö░ļĪ£ Ēś╣ņØĆ ļÅÖņŗ£ņŚÉ ļéśĒāĆļéśļŖö ņ¦łĒÖśĻĄ░ņØä ĒåĄņ╣ŁĒĢ£ļŗż[1]. ņØ┤ ņżæ Ļ░äņ¦łņØś ņä¼ņ£ĀĒÖöĻ░Ć Ļ░Ćņן ĒØöĒĢ£ Ēæ£ĒśäĒśĢ(phenotype)ņ£╝ļĪ£, ņØ┤ļĪ£ ņØĖĒĢśņŚ¼ ĒÖśņ×ÉļŖö ĒÅÉĒÖ£ļ¤ēņØś Ļ░Éņåī ļ░Å Ļ░ĆņŖż ĻĄÉĒÖśņØś ņןņĢĀļź╝ Ļ▓ĮĒŚśĒĢśļ®░ ņŗ¼ĒĢĀ Ļ▓ĮņÜ░ļŖö ĒśĖĒØĪ ļČĆņĀä ļ░Å ņé¼ļ¦ØĻ╣īņ¦Ć ņØ┤ļź┤Ļ▓ī ļÉ£ļŗż. ņĢĮļ¼╝, ņ£ĀĻĖ░ Ēś╣ņØĆ ļ¼┤ĻĖ░ļ¼╝ņ¦łņŚÉ ļīĆĒĢ£ ļģĖņČ£, ļ░®ņé¼ņäĀ ņ╣śļŻī, Ļ▓░ņ▓┤ņĪ░ņ¦üņ¦łĒÖś ļō▒ ņ£Āļ░£ ņøÉņØĖņØä ņĢī ņłś ņ׳ļŖö Ļ▓ĮņÜ░Ļ░Ć ņ׳Ļ│Ā, ņ×ÉņäĖĒĢ£ Ļ▓Ćņé¼ņŚÉņä£ļÅä ļ░£ļ│æ ņøÉņØĖņØ┤ ļ░ØĒśĆņ¦Ćņ¦Ć ņĢŖņØä ļĢīļÅä ņ׳ļŖöļŹ░, ņØ┤ļ¤¼ĒĢ£ Ļ▓ĮņÜ░ļź╝ ĒŖ╣ļ░£ņä▒ Ļ░äņ¦łņä▒ ĒÅÉļĀ┤(idiopathic interstitial pneumonia, IIP)ņØ┤ļØ╝ ņ¦Ćņ╣ŁĒĢśļ®░ ĻĘĖņżæ Ļ░Ćņן ņśłĒøäĻ░Ć ļČłļ¤ēĒĢ£ Ļ▓āņØ┤ ĒŖ╣ļ░£ņä▒ ĒÅÉņä¼ņ£Āņ”Ø(idiopathic pulmonary fibrosis, IPF)ņØ┤ļŗż[2]. IPFļŖö 65ņäĖ ņØ┤ņāüņØś ļģĖņØĖņŚÉņä£ ĒśĖļ░£ĒĢśĻ│Ā, ņłśĻ░£ņøöņŚÉņä£ ņłśļģäņŚÉ Ļ▒Ėņ│É ņ¦äĒ¢ēĒĢśļŖö ĒśĖĒØĪĻ│żļ×Ć, ļ¦łļźĖ ĻĖ░ņ╣© ļ░Å ņ▓Łņ¦äņŗ£ ņ¢æņĖĪ ĒÅÉĒĢśļČĆņØś ĒØĪĻĖ░ ņłśĒżņØīņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢśļ®░, ĒÖśņ×ÉļōżņØś ņżæĻ░ä ņāØņĪ┤ĻĖ░Ļ░äņØĆ ņ¦äļŗ© Ēøä 3-5ļģäņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[3]. ĒØĪņŚ░ņØĆ IPFņØś ņל ņĢīļĀżņĀĖ ņ׳ļŖö ņ£äĒŚśņØĖņ×ÉļĪ£, ĻĘĖ ļ░¢ņŚÉ ņ£ä-ņŗØļÅäņŚŁļźś, Epstein Barr virusļéś Hepatitis C virus ļō▒ņØś Ļ░ÉņŚ╝ ļ░Å ņ£ĀņĀäņ×É ļ│ĆņØ┤ ļō▒ļÅä ņ¦łļ│æ ļ░£ņāØĻ│╝ Ļ┤ĆļĀ© ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż.
2000ļģä IPFņØś ņ¦äļŗ©Ļ│╝ ņ╣śļŻīņŚÉ ļīĆĒĢ£ ņĀäļ¼ĖĻ░Ć ņØśĻ▓¼ņØä ĻĖ░ļ░śņ£╝ļĪ£ ĒĢ£ ņ▓½ ĻĄŁņĀ£ ņ¦Ćņ╣©ņØ┤ ļ░£Ēæ£ļÉ£ ņØ┤Ēøä[4], ņ×äņāü ņ”Øņāü, ļ░£ļ│æ ĻĖ░ņĀä, ņ×ÉņŚ░ Ļ▓ĮĻ│╝ņŚÉ ļīĆĒĢ£ ņŚ¼ļ¤¼ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ļōżņØä ĒåĄĒĢśņŚ¼ 2011ļģäņŚÉ ņ¦äļŗ©Ļ│╝ ņ╣śļŻīņŚÉ ļīĆĒĢ£ ĻĘ╝Ļ▒░ņżæņŗ¼(evidence-based)ņØś ņ¦äļŻī ņ¦Ćņ╣©ņØ┤ ļ░£Ēæ£ļÉśņŚłļŗż[5]. ĒĢśņ¦Ćļ¦ī ņØ┤Ēøä ņŚ░ĻĄ¼ļōżņŚÉņä£ ĻĖ░ņĪ┤ņØś ņ¦äļŻī ņ¦Ćņ╣©ņØ┤ ņŗżņĀ£ ņ¦äļŻīņŚÉ ņ׳ņ¢┤ņä£ņØś ņĀüņÜ®ņŚÉ ĒĢ£Ļ│äļź╝ ļ│┤ņ×äņŚÉ ļö░ļØ╝[6,7], ĻĖ░ņĪ┤ņØś IPFņØś ņ¦äļŗ© ĻĖ░ņżĆņØä ļ│┤ņÖäĒĢ£ ņāłļĪ£ņÜ┤ ņ¦äļŻī ņ¦Ćņ╣©ņØ┤ 2018ļģäņŚÉ ņä£ļĪ£ ļŗżļźĖ ĻĘĖļŻ╣ņŚÉņä£ ņĀ£ņŗ£ļÉśņŚłļŗż[3,8].
IPFņØś ļČłļ¤ēĒĢ£ ņśłĒøäļź╝ Ļ░ÉņĢłĒĢśļ®┤, ņ×äņāü ņØśņé¼Ļ░Ć ņĪ░ĻĖ░ņŚÉ ņĀĢĒÖĢĒĢ£ ņ¦äļŗ©ņØä ļé┤ļ”¼ļŖö Ļ▓āņØ┤ IPF ĒÖśņ×ÉņØś ņśłĒøäņŚÉ ļ¦żņÜ░ ņżæņÜöĒĢ£ļŹ░, ņØ┤ļŖö pirfenidone [7], nintedanib [9]Ļ│╝ Ļ░ÖņØĆ ņ¦łļ│æ ņ¦äĒ¢ē ņ¢ĄņĀ£ņŚÉ ņ£ĀĒÜ©ĒĢ£ ņĢĮļ¼╝ņØś Ļ░£ļ░£ļĪ£ ĻĘĖ ņżæņÜöņä▒ņØ┤ ļŹöņÜ▒ Ļ░ĢņĪ░ļÉśņŚłļŗż. ļ│Ė ņóģņäżņØĆ ņāłļĪ£ņÜ┤ ņ¦äļŻī ņ¦Ćņ╣©ņŚÉ ļö░ļźĖ IPFņØś ņ¦äļŗ© Ļ│╝ņĀĢņŚÉ ļīĆĒĢśņŚ¼ ĻĖ░ņłĀĒĢśĻ│Āņ×É ĒĢ£ļŗż.
ļ│Ė ļĪĀ
ņ¦äļŗ© Ļ│╝ņĀĢ
IPFĻ░Ć ņØśņŗ¼ļÉśļŖö ĒÖśņ×ÉņŚÉņä£ ļ│æļĀź ņ▓ŁņĘ©ņÖĆ ņØ┤ĒĢÖņĀü Ļ▓Ćņé¼ļź╝ ĒåĄĒĢśņŚ¼ ņ¦üņŚģņä▒ ĒÅÉņ¦łĒÖśņØ┤ļéś Ļ▓░ņ▓┤ņĪ░ņ¦üņ¦łĒÖś ļō▒ ņøÉņØĖ Ļ░Éļ│äņØ┤ Ļ░ĆļŖźĒĢ£ ņ¦łĒÖś ņŚ¼ļČĆļź╝ ĒīÉļŗ©ĒĢ£ļŗż. Ļ│ĀĒĢ┤ņāüļÅä ĒØēļČĆ ņĀäņé░ĒÖöļŗ©ņĖĄņ┤¼ņśü(high resolution computed tomography, HRCT)ņØä ņŗ£Ē¢ēĒĢśĻ│Ā, ņØ┤Ēøä ļŗżĒĢÖņĀ£ ĒåĀļĪĀ(multidisciplinary discussion, MDD)ņØä ĒåĄĒĢśņŚ¼ ĻĖ░Ļ┤Ćņ¦ĆĒÅÉĒżņäĖņ▓Ö Ļ▓Ćņé¼(bronchoalveolar lavage, BAL)ļéś ņÖĖĻ│╝ņĀü ĒÅÉņāØĻ▓Ć(surgical lung biopsy, SLB) ļō▒ņØś ņČöĻ░ĆņĀüņØĖ Ļ▓Ćņé¼ ņŗ£Ē¢ē ņŚ¼ļČĆļź╝ Ļ▓░ņĀĢĒĢ£ļŗż. ĒŖ╣ņ¦ĢņĀüņØĖ IPFņØś HRCT ņåīĻ▓¼ņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░(ĒåĄņāüĒśĢ Ļ░äņ¦łņä▒ ĒÅÉļĀ┤[usual interstitial pneumonia, UIP]ĒśĢ)ļŖö ņĪ░ņ¦ü Ļ▓Ćņé¼ ņŚåņØ┤ļÅä IPFļź╝ ņ¦äļŗ©ĒĢĀ ņłś ņ׳ņ£╝ļéś, ĻĘĖļĀćņ¦Ć ņĢŖņØä Ļ▓ĮņÜ░ļŖö ņĪ░ņ¦ü Ļ▓Ćņé¼ ļō▒ ņČöĻ░ĆņĀüņØĖ Ļ▓Ćņé¼ Ēøä ļŗżņŗ£ MDDļź╝ ĒåĄĒĢśņŚ¼ ņĄ£ņóģņĀüņ£╝ļĪ£ IPF ņ¦äļŗ©ņØä ĒĢśĻ▓ī ļÉ£ļŗż(Fig. 1).
ļ│æļĀź ņ▓ŁņĘ© ļ░Å ĻĖ░ļ│Ė Ļ▓Ćņé¼
Ļ│ĀļĀ╣ņ×É(ĒŖ╣Ē׳ 60ņäĖ ņØ┤ņāüņØś ļé©ņ×É)Ļ░Ć 6Ļ░£ņøö ņØ┤ņāü ņ¦äĒ¢ēļÉśļŖö ĒśĖĒØĪĻ│żļ×ĆņØ┤ļéś ļ¦łļźĖ ĻĖ░ņ╣©ņØä ĒśĖņåīĒĢśĻ│Ā, ņŗĀņ▓┤ ņ¦äņ░░ņŚÉņä£ Ļ│żļ┤ēņ¦Ć(clubbing) Ēś╣ņØĆ ĒØĪĻĖ░ņŗ£ ņłśĒżņØīņØ┤ ļōżļ”¼Ļ▒░ļéś, ĒØēļČĆ X-rayņŚÉņä£ ņ¢æĒÅÉĒĢśļČĆņØś ņ”ØĻ░ĆļÉ£ ņØīņśüņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ IPFļź╝ ņØśņŗ¼ĒĢ┤ņĢ╝ ĒĢ£ļŗż. ļéśņØ┤Ļ░Ć 40-60ņäĖļĪ£ ļ╣äĻĄÉņĀü ņĀŖņØĆ Ļ▓ĮņÜ░ņŚÉļÅä ĒÅÉņä¼ņ£Āņ”ØņØś Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳ļŗżļ®┤ IPFņØś Ļ░ĆļŖźņä▒ņØä ņŚ╝ļæÉņŚÉ ļæÉĻ│Ā ņ×ÉņäĖĒĢ£ ļ│æļĀź ņ▓ŁņĘ©Ļ░Ć ĒĢäņÜöĒĢśļŗż. ĒØĪņŚ░ļĀźĻ│╝ ņ¦üņŚģņä▒ ĒÅÉņ¦łĒÖśņØś ļ░░ņĀ£ļź╝ ņ£äĒĢśņŚ¼ ņ¦üņŚģļĀźņŚÉ ļīĆĒĢ£ ņ×ÉņäĖĒĢ£ ļ¼Ėņ¦äņØä ņŗ£Ē¢ēĒĢśĻ│Ā, Ļ│╝ļ»╝ņä▒ĒÅÉņןņŚ╝ ļō▒ņØś Ļ░ĆļŖźņä▒ņØä Ļ│ĀļĀżĒĢśņŚ¼ ĒÖśĻ▓ĮņŚÉ ļīĆĒĢ£ ņĀĢļ│┤(Ļ▒░ņŻ╝ĒĢśĻ▒░ļéś ĻĘ╝ļ¼┤ĒĢśļŖö Ļ││ņŚÉņä£ Ļ│░ĒīĪņØ┤ļéś ņāłņÖĆ ņןĻĖ░Ļ░ä ņĀæņ┤ēĒĢśņśĆļŖöņ¦Ć ļō▒)ļź╝ ņ¢╗ņ¢┤ņĢ╝ ĒĢ£ļŗż. ļśÉĒĢ£, ļ│ĄņÜ®ĒĢśļŖö ņĢĮļ¼╝ņØ┤ļéś ļ░®ņé¼ņäĀ ņ╣śļŻī ņŚ¼ļČĆ ļō▒ņŚÉ ļīĆĒĢ┤ņä£ļÅä ĒÖĢņØĖĒĢśņŚ¼ņĢ╝ ĒĢ£ļŗż. Ļ▓░ņ▓┤ņĪ░ņ¦üņ¦łĒÖś(connective tissue disease)ņØś ļÅÖļ░ś Ļ░ĆļŖźņä▒ņØä Ļ│ĀļĀżĒĢśņŚ¼ Ļ┤ĆņĀł ļ░Å Ēö╝ļČĆ ļō▒ Ļ┤ĆļĀ© ņ”ØņāüņŚÉ ļīĆĒĢ£ ļ│æļĀź ņ▓ŁņĘ©Ļ░Ć ĒĢäņÜöĒĢśĻ│Ā, Ļ┤ĆņĀł ļō▒ ĒÅÉ ņÖĖ ņ”Øņāü ņŚåņØ┤ ILDĻ░Ć ņäĀĒ¢ēĒĢśļŖö Ļ▓ĮņÜ░Ļ░Ć ņ׳ņ£╝ļ»ĆļĪ£ ļ¼┤ņ”ØņāüņØ┤ļØ╝ļÅä ĒĢŁĒĢĄĒĢŁņ▓┤(antinuclear antibody)ļéś ļźśļ¦łĒŗ░ņŖż ņØĖņ×É(rheumatoid factor) ļō▒ ņ×ÉĻ░ĆĒĢŁņ▓┤ Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢ£ļŗż[3].
Ļ│ĀĒĢ┤ņāüļÅä ĒØēļČĆ ņĀäņé░ĒÖöļŗ©ņĖĄņ┤¼ņśü(HRCT)
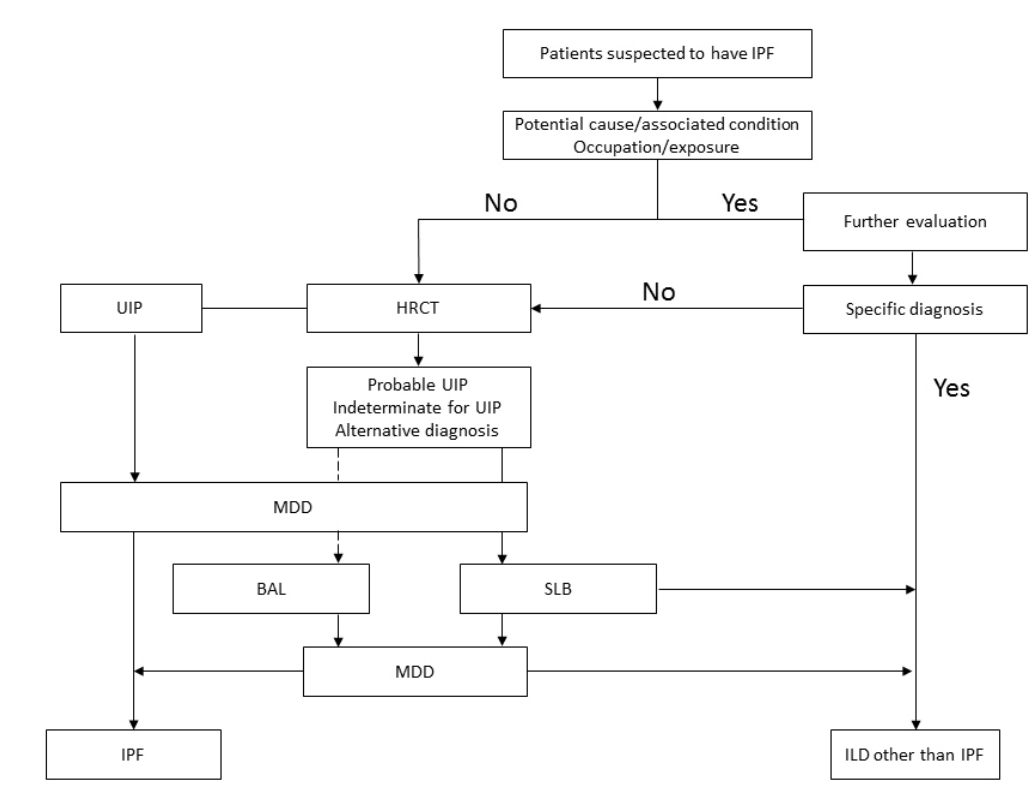
HRCTļŖö 2 mm ņØ┤ĒĢśņØś ņ¢ćņØĆ ļæÉĻ╗ś(thickeness)ļź╝ Ļ░¢ļŖö ņśüņāüņØä 1-2 cm Ļ░äĻ▓®ņ£╝ļĪ£ ņ┤¼ņśüĒĢśņŚ¼ ĒÅÉņŗżņ¦łņØś ļ│ĆĒÖöļź╝ ļ»╝Ļ░ÉĒĢśĻ▓ī ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŖö CT ņ┤¼ņśüĻĖ░ļ▓ĢņØ┤ļŗż[10]. Ļ│ĄĻĖ░Ļ▒Ėļ”╝(air trapping)ņØ┤ļéś ĒÅÉĒŚłĒāł(atelectasis) ļō▒ņØś ļÅÖļ░ś ņŚ¼ļČĆļź╝ ĒÖĢņØĖĒĢśĻĖ░ ņ£äĒĢśņŚ¼ ĒØĪĻĖ░ ļ¦É ņśüņāü ņÖĖņŚÉ ĒśĖĻĖ░ ļ¦ÉĻ│╝ ļ│ĄņÖĆņ£ä ņ×ÉņäĖ(prone position)ņŚÉņä£ļÅä ņ┤¼ņśüĒĢśļŖö Ļ▓āņØ┤ ļŗżļźĖ ILDļéś Ļ░Ćņä▒ļ│æņåī(pseudolesion) Ļ░Éļ│äņŚÉ ļÅäņøĆņØ┤ ļÉ£ļŗż[3] (Fig. 2).
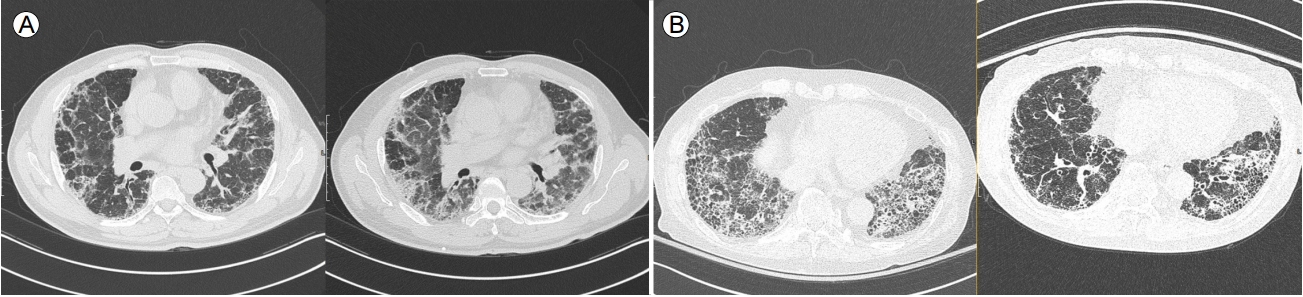
2011ļģäņŚÉ ļ░£Ēæ£ļÉ£ IPF ņ¦äļŻī ņ¦Ćņ╣©ņØĆ ņ▓ĀņĀĆĒĢ£ ļ│æļĀź ņ▓ŁņĘ©, ņ×ÉĻ░ĆĒĢŁņ▓┤ ļō▒ņØś Ļ▓Ćņé¼ņŚÉņä£ ņøÉņØĖņØ┤ ļ░ØĒśĆņ¦Ćņ¦Ć ņĢŖņØĆ IIP ĒÖśņ×ÉņŚÉņä£ HRCTļź╝ ļ©╝ņĀĆ ņŗ£Ē¢ēĒĢśļÅäļĪØ ĒĢśĻ│Ā, ĻĘĖ ņåīĻ▓¼ņŚÉ ļö░ļØ╝ 3Ļ░Ćņ¦Ć ņ£ĀĒśĢ(UIP pattern, possible UIP pattern, inconsistent UIP pattern)ņ£╝ļĪ£ ļČäļźśĒĢśņŚ¼, UIP patternņØĖ Ļ▓ĮņÜ░ļź╝ ņĀ£ņÖĖĒĢśĻ│ĀļŖö SLBļź╝ ĒĢśļÅäļĪØ ĻČīĻ│ĀĒĢśņśĆļŗż[5]. HRCT ņāü ĒÅÉĒĢśļČĆ, ļ│ĆņŚ░ļČĆ ņÜ░ņäĖņä▒ņ£╝ļĪ£ ļ┤ēņÖĆņ¢æĒÅÉ(honeycombing)ņÖĆ ļ¦ØņāüņØīņśü(reticular abnormality)ņØä ļÅÖļ░śĒĢśĻ│Ā, ļŗżļźĖ ņ¦łĒÖśņØä ņŗ£ņé¼ĒĢśļŖö ņåīĻ▓¼(ņä¼ņ£ĀĒÖö ļ│æļ│Ćļ│┤ļŗż ļ▓öņ£äĻ░Ć ļŹö ļäōņØĆ Ļ░äņ£Āļ”¼ņØīņśü[ground glass opacity], ĒÅÉĻ▓ĮĻ▓░[consolidation], ņżæ-ņāüņŚĮ ņ╣©ļ▓ö ļō▒)ņØ┤ ņŚåļŖö Ļ▓ĮņÜ░, ņśüņāüņØśĒĢÖņĀüņ£╝ļĪ£ UIP patternņ£╝ļĪ£ ļČäļźśĒĢśĻ│Ā, ņØ┤ļ¤¼ĒĢ£ Ļ▓ĮņÜ░ ļ│æļ”¼ ņåīĻ▓¼ņāü UIP patternĻ│╝ ņØ╝ņ╣śļÅäĻ░Ć ļ¦żņÜ░ ļåÆņØĆ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż[11]. ĻĖ░ņĪ┤ņØś ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö ņĢ×ņä£ņé┤ĒÄ┤ļ│Ė UIP patternņØś 4Ļ░Ćņ¦Ć ĒŖ╣ņ¦Ģ(ĒÅÉĒĢśļČĆ/ļ│ĆņŚ░ļČĆ ņÜ░ņäĖņä▒, ļ┤ēņÖĆņ¢æĒÅÉ, ļ¦ØņāüņØīņśü, ļŗżļźĖ ņ¦łĒÖśņØä ņŗ£ņé¼ĒĢśļŖö ņåīĻ▓¼ņØ┤ ņŚåņØī) ņżæ ļ┤ēņÖĆņ¢æĒÅÉļź╝ ņĀ£ņÖĖĒĢ£ ļéśļ©Ėņ¦Ć 3Ļ░Ćņ¦Ćļź╝ ļ¦īņĪ▒ĒĢ£ Ļ▓ĮņÜ░ļź╝ possible UIP patternņ£╝ļĪ£ ņĀĢņØśĒĢśņśĆļŖöļŹ░, Chung ļō▒[12]ņØĆ possible UIP patternņØä ņä¼ņ£ĀĒÖöņØś ņĀĢļÅäļĪ£ ļéśļłäņ¢┤ probable UIP (ĒÅÉĒĢśļČĆ, ļ│ĆņŚ░ļČĆ ņÜ░ņäĖņä▒, ļ¦ØņāüņØīņśü ņĪ┤ņ×¼, ļŗżļźĖ ņ¦łĒÖśņØä ņŗ£ņé¼ĒĢśļŖö ņåīĻ▓¼ņØ┤ ņŚåņØī)ņÖĆ indeterminate UIP pattern (CT ņāü ĒÅÉņä¼ņ£ĀĒÖöĻ░Ć ņĪ┤ņ×¼ĒĢśļéś definite, probable Ēś╣ņØĆ inconsistent with UIPļĪ£ ļČäļźśĒĢĀ ņĀĢļÅäļĪ£ ĒŖ╣ņ¦ĢņĀüņØ┤ņ¦Ć ņĢŖņØĆ Ļ▓ĮņÜ░)ņ£╝ļĪ£ ļéśļłäņ¢┤ ļ│æļ”¼ņĪ░ņ¦ü Ļ▓░Ļ│╝ļź╝ ļ╣äĻĄÉĒĢśņśĆĻ│Ā, probable UIP patternņŚÉņä£ ļ│æļ”¼ Ļ▓Ćņé¼ņāü UIP patternņØ┤ ļŹö ņ×ÉņŻ╝ Ļ┤Ćņ░░ļÉ©ņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż(82.4% vs. 54.2%; p= 0.01). ņØ┤ņŚÉ ļö░ļØ╝ 2018ļģä Ļ░£ņĀĢļÉ£ ņ¦Ćņ╣©ņŚÉņä£ļŖö ĒØēļČĆ HRCT ņåīĻ▓¼ņØä UIP, probable UIP, indeterminate for UIP, alternative diagnosisņØś 4Ļ░Ćņ¦ĆļĪ£ ļČäļźśĒĢśņśĆļŗż(Table 1, Fig. 3) [3,8].
ņÖĖĻ│╝ņĀü ĒÅÉņāØĻ▓Ć(SLB)
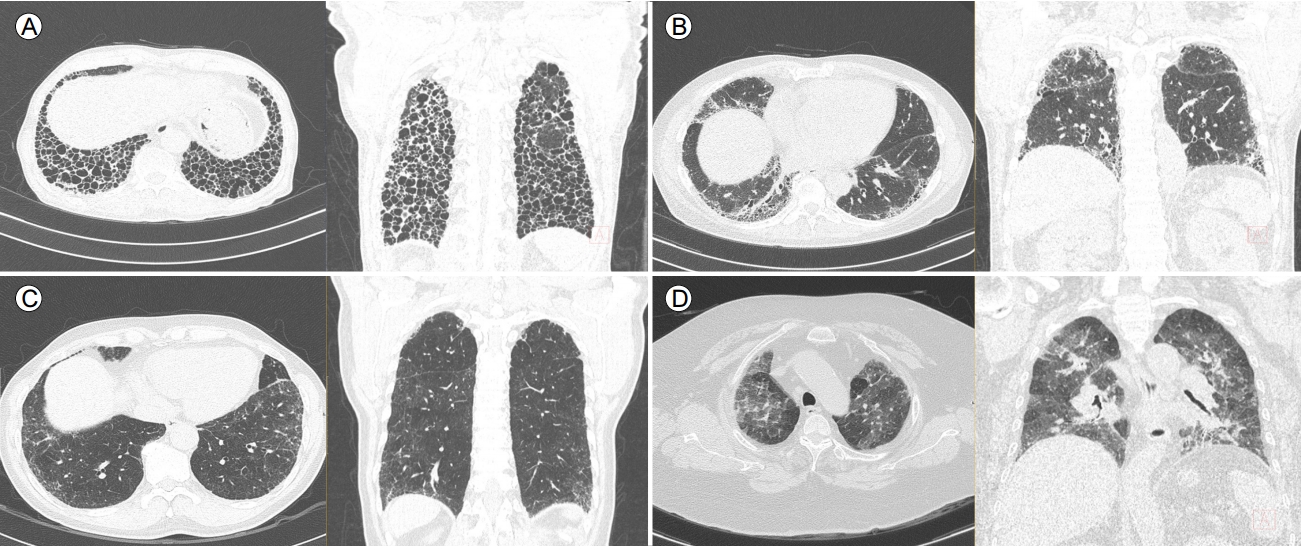
2018ļģä Ļ░£ņĀĢļÉ£ ņ¦äļŻī ņ¦Ćņ╣©ņŚÉ ļö░ļź┤ļ®┤ ņĪ░ņ¦ü Ļ▓Ćņé¼ ņåīĻ▓¼ļÅä ņśüņāüĻ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ UIP, probable UIP, indeterminate for UIP, alternative diagnosisņØś 4Ļ░Ćņ¦ĆļĪ£ ļČäļźśĒĢśņśĆļŗż(Table 2, Fig. 4). Ļ░ÖņØĆ ĒÖśņ×ÉņØś ņä£ļĪ£ ļŗżļźĖ ļæÉ ņŚĮņŚÉņä£ ļŗżļźĖ ņĪ░ņ¦üĒśĢņØ┤ Ļ┤Ćņ░░ļÉĀ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ(discordant UIP; ņ”ē ĒĢ£ ņŚĮņŚÉņä£ļŖö UIP pattern, ļŗżļźĖ ņŚĮņŚÉņä£ļŖö nonspecific interstitial pneumonia pattern), SLB ņŗ£Ē¢ē ņŗ£ ņĄ£ņåī 2Ļ││ ņØ┤ņāüņØś ĒÅÉņŚĮņŚÉņä£ ņĪ░ņ¦üņØä ņ¢╗ņ¢┤ņĢ╝ ĒĢ£ļŗż[3].
ĒÅÉņāØĻ▓Ć Ēøä ļ░£ņāØĒĢĀ ņłś ņ׳ļŖö ĒĢ®ļ│æņ”Øņ£╝ļĪ£ļŖö ĻĖēņä▒ņĢģĒÖö(6.1%), ņ¦ĆņåŹņĀüņØĖ Ļ│ĄĻĖ░ ļłäņČ£(6-12%), Ļ░ÉņŚ╝(6.5%), ņČ£Ēśł(0.2-0.8%) ļō▒ņØ┤ ņĢīļĀżņĀĖ ņ׳ļŗż[13]. Hutchinson ļō▒[14]ņØĆ 11ļģäĻ░ä 32,022ļ¬ģņØś SLBļź╝ ļ░øņØĆ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ņé¼ļ¦ØĻ│╝ ņŚ░Ļ┤ĆļÉ£ ņØĖņ×Éļź╝ ļČäņäØĒĢśņśĆĻ│Ā ļé©ņ×É, Ļ│ĀļĀ╣, ĻĖ░ņĀĆ ņ¦łĒÖśņØ┤ ļ¦ÄņØäņłśļĪØ ņé¼ļ¦ØņØ┤ ņ£ĀņØśĒĢśĻ▓ī ņ”ØĻ░ĆĒĢ©ņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż. ĒŖ╣Ē׳ 75ņäĖ ņØ┤ņāüņØĆ 45ņäĖ ļ»Ėļ¦īņŚÉ ļ╣äĒĢśņŚ¼ 4.5ļ░░ ņé¼ļ¦Ø ņ£äĒŚśļÅäĻ░Ć ņ”ØĻ░ĆĒĢśņśĆļŗż. ļśÉ ņĢłņĀĢņŗ£ Ēś╣ņØĆ ņÜ┤ļÅÖņŗ£ ņé░ņåīļź╝ ĒĢäņÜöļĪ£ ĒĢśļŖö Ļ▓ĮņÜ░ļŖö ņłśņłĀ Ēøä ņé¼ļ¦Ø, ĒÅÉļĀ┤, ņ¦ĆņåŹņĀüņØĖ Ļ│ĄĻĖ░ ļłäņČ£ ļō▒ņØś ņŻ╝ņÜö ĒĢ®ļ│æņ”ØņØ┤ ļ░£ņāØĒĢĀ ĒÖĢļźĀņØ┤ ņ”ØĻ░ĆĒĢ£ļŗż. LoCicero [15]ņØś ņŚ░ĻĄ¼ņŚÉņä£ļÅä ĒÅÉņāØĻ▓Ćņŗ£ ņé░ņåī ņÜöĻĄ¼ļ¤ēņØ┤ ņŚåņØä Ļ▓ĮņÜ░ ņé¼ļ¦ØļźĀņØĆ 4%, ņłśņłĀ ņĀä ņé░ņåīĻ░Ć ĒĢäņÜöĒĢ£ Ļ▓ĮņÜ░ļŖö 6% ĻĘĖļ”¼Ļ│Ā ĒśĖĒØĪļČĆņĀäņ£╝ļĪ£ ĻĖ░Ļ│ä ĒÖśĻĖ░Ļ░Ć ĒĢäņÜöĒĢ£ ĒÖśņ×ÉņŚÉņä£ļŖö 75%ļĪ£ ļ│┤Ļ│ĀĒĢśņśĆļŗż. ņĪ░ņ¦ü Ļ▓Ćņé¼ļź╝ ĒĢĀ ļŗ╣ņŗ£ ĻĖēņä▒ņĢģĒÖöņØś ļ│æļĀźļÅä ņĢīļĀżņ¦ä ņ£äĒŚśņØĖņ×ÉļĪ£ņä£, Park ļō▒[16]ņØĆ SLBļź╝ ņŗ£Ē¢ēļ░øņØĆ 200ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ ĻĖēņä▒ņĢģĒÖöņŗ£ ņĪ░ņ¦ü Ļ▓Ćņé¼ļź╝ ļ░øņØĆ Ļ▓ĮņÜ░ ņé¼ļ¦ØļźĀ(28.6%)ņØ┤ ĻĘĖļĀćņ¦Ć ņĢŖņØĆ Ļ▓ĮņÜ░(3.0%)ņŚÉ ļ╣äĒĢśņŚ¼ ņ£ĀņØśĒĢśĻ▓ī ņ”ØĻ░ĆĒĢśĻ│Ā, ĒÅÉĒÖĢņé░ļŖź(diffusing capacity for carbon monoxide)ņØ┤ ļé«ņØäņłśļĪØ ņĪ░ņ¦ü Ļ▓Ćņé¼ Ēøä ĒĢ®ļ│æņ”ØņØ┤ ļ░£ņāØĒĢĀ ņ£äĒŚśņØ┤ ļåÆņØī(odds ratio, 0.959; 95% confidence interval, 0.930-0.989; p= 0.007)ņØä ļ│┤ņŚ¼ņŻ╝ņŚłļŗż. ĻĘĖ ņÖĖņŚÉ Ļ│ĀļĀ╣, ļ®┤ņŚŁņĀĆĒĢś ņāüĒā£, ņżæņ”ØņØś ĒÅÉĻ│ĀĒśłņĢĢņØ┤ ņ׳ņØä Ļ▓ĮņÜ░ ņłśņłĀ Ēøä ņé¼ļ¦ØļźĀņØ┤ ņ”ØĻ░ĆĒĢśņŚ¼ ņŻ╝ņØśĻ░Ć ĒĢäņÜöĒĢśļŗż[15,17-19].
2011ļģä ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ SLBņŚÉ ļīĆĒĢ┤ņä£ ļ╣äļööņśż ĒØēĻ░ĢĻ▓Į ņłśņłĀ(video assisted thoracic surgery, VATS)Ļ│╝ Ļ░£ĒØēņłĀ(open lung thoracotomy, OLB)ņØä Ļ░ÖņØĆ ņłśņżĆņ£╝ļĪ£ ņåīĻ░£ĒĢśņśĆņ£╝ļéś, Ļ░£ņĀĢļÉ£ ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö VATSļź╝ ņČöņ▓£ĒĢśņśĆļŗż. 41ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ VATSņÖĆ OLBņØś ĒÜ©ņÜ®ņä▒ņØä ļ╣äĻĄÉĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£, VATS ņŗ£Ē¢ēņŗ£ ņłśņłĀ ņŗ£Ļ░ä(45.3 ┬▒ 12.2ļČä vs. 55.6 ┬▒ 11.2ļČä)Ļ│╝ ņ×ģņøÉ ĻĖ░Ļ░ä(5.5 ┬▒ 1.3ņØ╝ vs. 7.1 ┬▒ 2.3ņØ╝)ņØ┤ OLBņŚÉ ļ╣äĒĢśņŚ¼ ņżäņ¢┤ļōżņŚłņØīņØä ļ│┤ņŚ¼ņŻ╝ņŚłĻ│Ā[20], ņé¼ļ¦ØļźĀļÅä ņ£ĀņØśĒĢśĻ▓ī ļé«ņĢśļŗż(1.18% vs. 2.29%, p< 0.001) [14].
ņĄ£ĻĘ╝ ĻĖ░Ļ┤Ćņ¦Ć ļé┤ņŗ£Ļ▓ĮņØä ņØ┤ņÜ®ĒĢ£ ļāēļÅÖĒÅÉņāØĻ▓Ć(bronchoscopic lung cryobiopsy) ņŗ£ņłĀņØ┤ ILDņØś ņ¦äļŗ©ņØä ņ£äĒĢ£ ņĪ░ņ¦ü Ļ▓Ćņé¼ ļ░®ļ▓Ģņ£╝ļĪ£ ņŗ£ļÅäļÉśĻ│Ā ņ׳ļŗż[21,22]. 117ļ¬ģņØś ILD ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ļāēļÅÖĒÅÉņāØĻ▓ĆĻ│╝ VATSļź╝ ļ╣äĻĄÉĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£, ņĪ░ņ¦üņØś ņĀüĒĢ®ņä▒(100% vs. 100%)ņØ┤ļéś ILDņØś ņ¦äļŗ©ņ£©(91% vs. 98%, p= 0.71) ļ░Å ņŗ£ņłĀ Ēøä ņé¼ļ¦ØļźĀ(1.7% vs. 3.4%)ņØ┤ ļæÉ ļ░®ļ▓Ģ Ļ░ä ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć ņŚåļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż[22]. ĒĢśņ¦Ćļ¦ī Ļ░ü ĻĖ░Ļ┤Ćļ│äļĪ£ ņłÖļĀ©ļÅäĻ░Ć ļŗżļź┤Ļ│Ā, ņŗ£ņłĀ Ļ│╝ņĀĢņØ┤ Ēæ£ņżĆĒÖöļÉśņ¢┤ ņ׳ņ¦Ć ņĢŖņĢä Ļ░£ņĀĢļÉ£ ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö ņĢäņ¦ü ILDļź╝ ņ¦äļŗ©ĒĢśļŖö ļ░®ļ▓Ģņ£╝ļĪ£ ĻČīĻ│ĀĒĢśĻ│Ā ņ׳ņ¦Ć ņĢŖļŗż[3].
ņÖĖĻ│╝ņĀü ĒÅÉņāØĻ▓Ć(SLB)ņØś ņĀüņØæņ”Ø
2011 American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Society (ALAT) ņ¦äļŻī ņ¦Ćņ╣©Ļ│╝ ļÅÖņØ╝ĒĢśĻ▓ī 2018ļģä ņ¦Ćņ╣©ņŚÉņä£ļÅä HRCT ņāü ņĀäĒśĢņĀüņØĖ UIPĒśĢņØä ļ│┤ņØ┤ņ¦Ć ņĢŖļŖö Ļ▓ĮņÜ░ļŖö SLBļź╝ Ļ│ĀļĀżĒĢśļÅäļĪØ ĒĢśņśĆļŗż. ĻĘĖļ¤¼ļéś 2011ļģä ņ¦Ćņ╣©ņØ┤ ļ░£Ēæ£ļÉ£ ņØ┤Ēøä HRCTņÖĆ ņĪ░ņ¦ü ņåīĻ▓¼ņØś ņØ╝ņ╣śņ£©ņŚÉ Ļ┤ĆĒĢ£ ņŚ░ĻĄ¼ļōżņØ┤ ņ¦äĒ¢ēļÉśņŚłĻ│Ā, ĒØēļČĆ HRCT ņāü ļ┤ēņÖĆņ¢æĒÅÉĻ░Ć ņŚåļŹöļØ╝ļÅä ĒØēļ¦ēĒĢśņŚÉ ļ¦ØņāüņØīņśüĻ│╝ Ļ▓¼ņØĖņä▒ ĻĖ░Ļ┤Ćņ¦ĆĒÖĢņןņ”ØņØä ļÅÖļ░śĒĢ£ Ļ▓ĮņÜ░ ņĪ░ņ¦ü Ļ▓Ćņé¼ņŚÉņä£ UIPĻ░Ć ņ¦äļŗ©ļÉĀ ĒÖĢļźĀņØ┤ ļåÆņØī(82-94%)ņØ┤ ĒÖĢņØĖļÉśņŚłļŗż[11,12]. ĒŖ╣Ē׳ 60ņäĖ ņØ┤ņāü ļé©ņ×É, ĒØĪņŚ░ļĀź ļō▒ņØś ņ×äņāüņ¢æņāüņØä ļ¦īņĪ▒ĒĢśļŖö Ļ▓ĮņÜ░ņŚÉļŖö ņØ╝ņ╣śļÅäĻ░Ć ļŹö ļåÆņĢäņ¦ÉņØä ĻĘ╝Ļ▒░ļĪ£[23], 2018ļģä Fleishner ĻĘĖļŻ╣ņŚÉņä£ļŖö IPFņØś ņ×äņāüņāüņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉņŚÉņä£ ĒØēļČĆ CT ņāü probable UIPĒśĢņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ SLB ņŚåņØ┤ļÅä IPFļź╝ ņ¦äļŗ©ĒĢĀ ņłś ņ׳ļŗżĻ│Ā ņĀ£ņŗ£ĒĢśņśĆļŗż[8]. ĒĢśņ¦Ćļ¦ī ņØ┤ņ¢┤ ļ░£Ēæ£ļÉ£ 2018ļģä ATS/ERS/JRS/ALAT ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö ĒØēļČĆ CT Ļ▓Ćņé¼ ņāü probable UIP ņåīĻ▓¼ņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ SLBļź╝ ņŗ£Ē¢ēĒĢśļÅäļĪØ ĻČīņ£ĀĒĢśņśĆļŗż(conditional recommendation) [3]. ļ╣äļĪØ Ēæ£ļ®┤ņĀüņ£╝ļĪ£ļŖö ĒØēļČĆ CT ņāü probable UIP ņåīĻ▓¼ņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉņŚÉ ļīĆĒĢ┤ņä£ ļæÉ ņ¦äļŻī ņ¦Ćņ╣©ņØś Ļ▓¼ĒĢ┤Ļ░Ć ļŗżļźĖ Ļ▓āņ▓śļ¤╝ ļ│┤ņØ┤ņ¦Ćļ¦ī, ATS/ERS/JRS/ALAT ņ¦äļŻī ņ¦Ćņ╣©ņØĆ conditional recommendationņ£╝ļĪ£ņä£, HRCTņŚÉņä£ probable UIP ņåīĻ▓¼ņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉļōżņØś ļŗżņłś(Ōēź 50%)ļŖö SLBĻ░Ć ĒĢäņÜöĒĢśņ¦Ćļ¦ī ņØ╝ļČĆ(<50%) ĒÖśņ×É(ņśłļź╝ ļōżļ®┤ ņ×äņāüņĀüņ£╝ļĪ£ IPFņØ╝ Ļ░ĆļŖźņä▒ņØ┤ ļåÆņØĆ)ņŚÉņä£ļŖö SLBĻ░Ć ĒĢäņÜöĒĢśņ¦Ć ņĢŖņØä ņłś ņ׳ļŗżļØ╝Ļ│Ā ĒĢ┤ņäØĒĢĀ ņłś ņ׳ļŗżļŖö ņĀÉņŚÉņä£ ļæÉ ņ¦Ćņ╣©ņØ┤ ņØśļÅäĒĢśļŖö ļ░öļŖö ļÅÖņØ╝ĒĢśļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż[24,25].
ĻĖ░Ļ┤Ćņ¦ĆĒÅÉĒżņäĖņ▓Ö Ļ▓Ćņé¼
ĻĖ░Ļ┤Ćņ¦ĆĒÅÉĒżņäĖņ▓Ö Ļ▓Ćņé¼(BAL)ļŖö ĻĖ░Ļ┤Ćņ¦Ć ļé┤ņŗ£Ļ▓ĮņØä ņØ┤ņÜ®ĒĢśņŚ¼ ĻĖ░ļÅäļé┤ ņ┤Ø 100-300 mLņØś ņāØļ”¼ņŗØņŚ╝ņłśļź╝ 3-5ĒÜī ļéśļłäņ¢┤ ņŻ╝ņ×ģ Ēøä ĒÜīņłśĒĢśņŚ¼ ļ¦Éņ┤ł ĻĖ░ļÅäņÖĆ ĒÅÉĒżņŚÉņä£ Ļ▓Ćņ▓┤ļź╝ ņ¢╗ļŖö Ļ▓Ćņé¼ļ░®ļ▓ĢņØ┤ļŗż[26]. ĒÜīņłśļÉ£ ņäĖņ▓ÖņĢĪņØĆ ņŻ╝ņ×ģļÉ£ ņ¢æņØś ņĄ£ņåī 30% ņØ┤ņāüņØ┤ ļÉśņ¢┤ņĢ╝ ĒĢśļ®░, ĻĘĖ ņØ┤ĒĢśņØ╝ Ļ▓ĮņÜ░ ņäĖĒż ļČäņ£©(differential counts)ņØä ĒĢ┤ņäØĒĢśļŖöļŹ░ ņŻ╝ņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż.
IPFņØś ĒÖśņ×ÉņŚÉņä£ BAL ņäĖņ▓ÖņĢĪ ņåīĻ▓¼ņØĆ ņĀĢņāüņØĖ Ēś╣ņØĆ ļŗżļźĖ ĒÅÉņ¦łĒÖśņŚÉ ļ╣äĒĢśņŚ¼ ĒśĖņżæĻĄ¼ņØś ļ╣äņ£©ņØ┤ ņ”ØĻ░ĆĒĢśņ¦Ćļ¦ī, Ļ▓░ņ▓┤ņĪ░ņ¦üĻ┤ĆļĀ© ĒÅÉņ¦łĒÖśņØ┤ļéś ļŗżļźĖ ņ¦łĒÖśņŚÉņä£ļÅä ņ£Āņé¼ĒĢ£ ņåīĻ▓¼ņØä ļ│┤ņØ╝ ņłś ņ׳ņ¢┤, 2011ļģä ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö IPF ņ¦äļŗ©ņØä ņ£äĒĢśņŚ¼ BALņØä ņŗ£Ē¢ēĒĢśļÅäļĪØ ĻČīĻ│ĀĒĢśņ¦Ć ņĢŖņĢśļŗż[5]. ĻĘĖļ¤¼ļéś IPFĻ░Ć ņĢäļŗī ļŗżļźĖ ņ¦łĒÖśņØä Ļ░Éļ│äĒĢśļŖöļŹ░ ļÅäņøĆņØ┤ ļÉĀ ņłś ņ׳ļŖöļŹ░, BAL ņäĖĒżļČäĒÜŹņŚÉņä£ ĒśĖņé░ĻĄ¼Ļ░Ć 25% ņØ┤ņāüņØ┤ļ®┤ ĒśĖņé░ĻĄ¼ņä▒ ĒÅÉļĀ┤(eosinophilic pneumonia)ņŚÉ ņ¦äļŗ©ņĀüņØ┤Ļ│Ā[26], ņ£Āņ£Īņóģņ”Ø(sarcoidosis)ņØś Ļ▓ĮņÜ░ ļ│┤ĒåĄ ļ”╝ĒöäĻĄ¼Ļ░Ć 15% ņØ┤ņāüņØ┤Ļ│Ā CD4/CD8 ļ╣äņ£©ņØ┤ 4 ņØ┤ņāüņ£╝ļĪ£ ļåÆļŗż. ļö░ļØ╝ņä£ ĒØēļČĆ CTņŚÉņä£ UIP patternņØ┤ ņĢäļŗī Ļ▓ĮņÜ░ ņāüĻĖ░ ņ¦łĒÖś ļō▒ņØä Ļ░Éļ│äĒĢĀ ļ¬®ņĀüņ£╝ļĪ£ BAL ņäĖĒżļČäĒÜŹĻ│╝ CD4/CD8 ļ╣äņ£©ņØä ĒÖĢņØĖĒĢśļŖö Ļ▓āņØ┤ ĻČīĻ│Ā ņé¼ĒĢŁņ£╝ļĪ£ ņāłļĪŁĻ▓ī ņČöĻ░ĆĻ░Ć ļÉśņŚłļŗż[3].
ļŗżĒĢÖņĀ£ ņ¦äļŗ©ņØś ņżæņÜöņä▒
IPF ņ¦äļŗ©ņØś Ēæ£ņżĆņØĆ ļŗżĒĢÖņĀ£ ĒåĀļĪĀņØä ĒåĄĒĢ£ ņ¦äļŗ©ņØ┤ļŗż[3]. Flaherty ļō▒[27]ņØĆ IIPņØś ņ¦äļŗ© Ļ│╝ņĀĢņŚÉņä£ ņ×äņāü, ļ│æļ”¼, ņśüņāü ņĀĢļ│┤ņØś ĒåĄĒĢ®Ļ│╝ Ļ░ü ļČäņĢ╝ ņĀäļ¼ĖĻ░Ć Ļ░ä ĒåĀļĪĀņØä ĒåĄĒĢśņŚ¼ ņ¦äļŗ©ņØś ņØ╝ņ╣śļÅä(interobserver agreement)ņÖĆ ņŗĀļó░ļÅäļź╝ ļåÆņØ╝ ņłś ņ׳ņØīņØä ļ│┤ņŚ¼ņŻ╝ņŚłļŗż. Chaudhuri ļō▒[28]ņØĆ ņ¦ĆņŚŁ ļ│æņøÉņØś ĒśĖĒØĪĻĖ░ ņĀäļ¼ĖņØśļĪ£ļČĆĒä░ MDDļź╝ ņØśļó░ļ░øņØĆ 318ļ¬ģņØś ILD ņ”ØļĪĆļź╝ ļČäņäØĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£, ņĄ£ņ┤ł ņ¦äļŗ©ņØ┤ IPFņśĆļŹś 107ļ¬ģ ņżæ ņĀłļ░śņØś ĒÖśņ×ÉņŚÉņä£ MDD Ēøä ņ¦äļŗ©ņØ┤ ļ│ĆĻ▓ĮļÉśņŚłņ£╝ļ®░, IPFļź╝ ņĀ£ņÖĖĒĢ£ ļŗżļźĖ ILD ĒÖśņ×É 136ļ¬ģļÅä 33%ņŚÉņä£ ņ¦äļŗ©ņØ┤ ļ│ĆĻ▓ĮļÉśņŚłņØīņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż. Jo ļō▒[29]ļÅä ĒśĖĒØĪĻĖ░ ņĀäļ¼ĖņØśļĪ£ļČĆĒä░ ņØśļó░ļ░øņØĆ 90ļ¬ģņØś ILD ĒÖśņ×É ņżæ MDDļź╝ ĒåĄĒĢśņŚ¼ 53%ņŚÉņä£ ņ¦äļŗ©ņØ┤ ļ│ĆĻ▓ĮļÉśņŚłĻ│Ā, ņ╣śļŻī ļ░®ņ╣©ņŚÉ ņ׳ņ¢┤ņä£ļÅä ĒĢŁņä¼ņ£ĀĒÖöņĀ£ņØś ņé¼ņÜ®(3% vs. 21%, p< 0.01)ņØ┤ļéś ņé░ņåī ņ╣śļŻī(6% vs. 10%, p= 0.046) ļō▒ņŚÉ ņ׳ņ¢┤ ņ£ĀņØśĒĢ£ ļ│ĆĒÖöĻ░Ć ņ׳ņØīņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż. ņØ┤ļ¤¼ĒĢ£ Ļ▓░Ļ│╝ļōżņØĆ MDDĻ░Ć ņ×äņāü ņ¦äļŻīņŚÉ ņ׳ņ¢┤ņä£, ņ¦äļŗ©ļ┐Éļ¦ī ņĢäļŗłļØ╝ ĒÖśņ×ÉņØś ņ╣śļŻī ļ░®ņ╣©ņØä Ļ▓░ņĀĢĒĢśĻ│Ā ņśłĒøäļź╝ ĒīÉļŗ©ĒĢśļŖö ļŹ░ņŚÉļÅä ņżæņÜöĒĢ£ ņśüĒ¢źņØä ņżīņØä ņŗ£ņé¼ĒĢ┤ ņżĆļŗż.
2018ļģä Ļ░£ņĀĢļÉ£ ņ¦äļŻī ņ¦Ćņ╣©ņŚÉņä£ļŖö MDDņØś ņżæņÜöņä▒ņØä ļŹöņÜ▒ Ļ░ĢņĪ░ĒĢśņŚ¼ ĒØēļČĆ HRCT ņĀĢļ│┤ļź╝ ņ¢╗ņØĆ Ēøä MDDļź╝ ņŗ£Ē¢ēĒĢśņŚ¼ BALņØ┤ļéś SLB ļō▒ ņČöĻ░ĆņĀüņØĖ Ļ▓Ćņé¼ ņŗ£Ē¢ē ņŚ¼ļČĆļź╝ Ļ▓░ņĀĢĒĢśļÅäļĪØ ĒĢśņśĆĻ│Ā, ņāüĻĖ░ Ļ▓Ćņé¼ Ļ▓░Ļ│╝ļōżņØä ĒåĀļīĆļĪ£ ļŗżņŗ£ ĒĢ£ļ▓ł MDDļź╝ ĒĢĀ Ļ▓āņØä ĻČīņ£ĀĒĢśņśĆļŗż(Fig. 1). ĒĢśņ¦Ćļ¦ī ņĀäĒśĢņĀüņØĖ IPFņØś ņ×äņāü-ņśüņāüņāüņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ņŚÉļŖö ļ░śļō£ņŗ£ MDDĻ░Ć ĒĢäņÜöĒĢśņ¦ĆļŖö ņĢŖļŗż[3]. Fleischner ĻĘĖļŻ╣ļÅä ņĪ░ņ¦ü Ļ▓Ćņé¼ ņŗ£Ē¢ē ņŚ¼ļČĆļź╝ Ļ▓░ņĀĢĒĢśĻ▒░ļéś, ņĪ░ņ¦ü Ļ▓Ćņé¼ Ēøä ņ×äņāü/ņśüņāü/ļ│æļ”¼ ņåīĻ▓¼ņØä ļ”¼ļĘ░ĒĢĀ ļĢī, ņ¦łļ│æņØś Ļ▓ĮĻ│╝Ļ░Ć ņĢīļĀżņ¦ä Ļ▓āĻ│╝ ļŗ¼ļØ╝ ņ×¼ĒÅēĻ░ĆĻ░Ć ĒĢäņÜöĒĢĀ Ļ▓ĮņÜ░ņŚÉ MDDļź╝ ņČöņ▓£ĒĢśņśĆļŗż[8]. MDDņØś ĻĄ¼ņä▒ņØĆ Ļ▓ĮĒŚśņØ┤ ļ¦ÄņØĆ ņ×äņāü, ņśüņāü, ļ│æļ”¼ņĀäļ¼ĖĻ░Ćļź╝ ĻĖ░ļ│Ėņ£╝ļĪ£, ĒĢäņÜöņŗ£ ĒÖśĻ▓Į, ņ£ĀņĀä, ļźśļ¦łĒŗ░ņŖżņ¦łĒÖś ņĀäļ¼ĖĻ░ĆĻ░Ć ņ░ĖņŚ¼ĒĢśļŖö Ļ▓āņØ┤ ĻČīĻ│ĀļÉ£ļŗż. ĒĢśņ¦Ćļ¦ī ņ░ĖņŚ¼ ņØĖņøÉņØä ņ¢┤ļ¢╗Ļ▓ī ĻĄ¼ņä▒ĒĢĀ Ļ▓āņØĖņ¦Ć, ĻĘĖļ”¼Ļ│Ā MDD ņŗ£Ē¢ē ņŻ╝ĻĖ░ ļō▒ņŚÉ ļīĆĒĢ£ Ļ▓░ņĀĢņØĆ Ļ░ü ņä╝Ēä░ņØś ņ¦äļŻī ņŚ¼Ļ▒┤ņŚÉ ļ¦×Ļ▓ī ņŗ£Ē¢ēļÉĀ ņłś ņ׳ļŗż.
Ļ▓░ ļĪĀ
IPFļŖö ņ¦äļŗ© Ēøä ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņ¦äĒ¢ēĒĢśņŚ¼ ļåÆņØĆ ņé¼ļ¦ØļźĀņØä ļ│┤ņØ┤ļŖö ņ¦łĒÖśņ£╝ļĪ£, IPFĻ░Ć ņØśņŗ¼ļÉśļŖö ĒÖśņ×ÉņØś ņĪ░ĻĖ░ ļ░£Ļ▓¼Ļ│╝ ņĀĢĒÖĢĒĢ£ ņ¦äļŗ© ļ░Å ņĀüņĀłĒĢ£ ņ╣śļŻīĻ░Ć ļ¦żņÜ░ ņżæņÜöĒĢśļŗż. Ļ░£ņĀĢļÉ£ IPF ņ¦äļŗ© ņ¦Ćņ╣©ņØĆ ĒØēļČĆ HRCT ņåīĻ▓¼ņāü UIPĒśĢņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░ ņÖĖņŚÉļÅä, ņ×äņāüņĀüņ£╝ļĪ£ IPFĻ░Ć ņØśņŗ¼ļÉśļŖö ĒÖśņ×É(working diagnosis of IPF)ņŚÉņä£ SLB ņŚåņØ┤ļÅä IPFļź╝ ņ¦äļŗ©ĒĢĀ ņłś ņ׳ļŖö ņŚ¼ņ¦Ćļź╝ ļæÉņŚłļŗż. ņØ┤ļ¤¼ĒĢ£ ņØśņé¼Ļ▓░ņĀĢņØĆ MDDļź╝ ĒåĄĒĢśņŚ¼ ņ¦äĒ¢ēļÉśņ¢┤ņĢ╝ ĒĢśļ®░, ņØ┤ļź╝ ĒåĄĒĢśņŚ¼ ņ╣©ņŖĄņĀü ņ¦äļŗ© ļ░®ļ▓ĢņØ┤ ĒĢäņÜöĒĢ£ ĒÖśņ×Éļź╝ ņäĀļ│äĒĢśņŚ¼ ļČłĒĢäņÜöĒĢśĻ▓ī ļ░£ņāØĒĢĀ ņłś ņ׳ļŖö ņłśņłĀ Ļ┤ĆļĀ© ĒĢ®ļ│æņ”ØņØä ņżäņØ┤Ļ│Ā ņĀĢĒÖĢĒĢ£ ņ¦äļŗ©ņØä ļé┤ļ”¼ļŖöļŹ░ ĻĖ░ņŚ¼ĒĢĀ Ļ▓āņ£╝ļĪ£ ņśłņāüĒĢ£ļŗż.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






