


м„ң лЎ
к°ҖмЎұм„ұ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқ(familial hypercholesterolemia, FH; ліё л…јл¬ём—җм„ңлҠ” лӢӨлҘё м–ёкёүмқҙ м—Ҷмңјл©ҙ мқҙнҳ•м ‘н•© нҳ•нғңлҘј м§Җм№ӯн•ңлӢӨ)мқҖ мғҒм—јмғүмІҙ мҡ°м„ұмқё лӢЁмқјмң м „мһҗ мң м „ м§Ҳнҷҳ мӨ‘ м ңмқј нқ”н•ҳлӢӨ. мқёкө¬ 200-500лӘ…лӢ№ н•ң лӘ…кјҙлЎң ліҙкі лҗҳм§Җл§Ң[1,2], м§Җм—ӯм Ғмқё нҺём°Ёк°Җ мһҲмқ„ мҲҳ мһҲлӢӨ. лӢӨлҘё лӮҳлқјмҷҖ 비мҠ·н• кІғмңјлЎң м¶”м •лҗҳм§Җл§Ң, н•ңкөӯм—җм„ң м •нҷ•н•ң мң лі‘лҘ мқҖ мһҳ м•Ңл Өм§Җм§Җ м•Ҡм•ҳлӢӨ. н•ңкөӯм—җм„ң 1980л…„лҢҖл¶Җн„° FH нҷҳмһҗ мӮ¬лЎҖк°Җ ліҙкі лҗҳм—ҲлӢӨ[3,4].
лі‘мқём„ұ ліҖмқҙк°Җ л°ңкІ¬лҗҳлҠ” лҢҖн‘ңм Ғ мң м „мһҗлҠ” LDLR, APOB, PCSK9мқёлҚ°, к°Ғ мң м „мһҗм—җ мғқкё°лҠ” лҸҢм—°ліҖмқҙлҠ” лӢӨм–‘н•ҳлӢӨ. л“ңл¬јкІҢ лӢӨлҘё мң м „мһҗм—җ нқ¬к·Җн•ң лі‘мқём„ұ ліҖмқҙк°Җ ліҙкі лҗҳкё°лҸ„ н•ңлӢӨ. н•ңнҺё FHм—җ л§һлҠ” н‘ңнҳ„нҳ•мқҙ мһҲлҠ” нҷҳмһҗ мӨ‘ мғҒлӢ№мҲҳм—җм„ң лі‘мқём„ұ ліҖмқҙк°Җ л°ңкІ¬лҗҳм§Җ м•ҠлҠ”лӢӨ. л”°лқјм„ң мқҙ м§Ҳнҷҳмқҙ мң м „м„ұмһ„м—җлҸ„ л¶Ҳкө¬н•ҳкі , 진лӢЁмқ„ н•ҳлҠ” лҚ°лҠ” мһ„мғҒ진лӢЁкё°мӨҖмқҙ мӨ‘мҡ”н•ҳлӢӨ. м„ёкі„м ҒмңјлЎң нҶөмқјлҗң 진лӢЁкё°мӨҖмқҖ м—Ҷмңјл©°, лӮҳлқјл§ҲлӢӨ лӘҮ к°Җм§Җ лӢӨлҘё кё°мӨҖмқ„ м“ҙлӢӨ. мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„лҠ” мөңкі 10л°°к№Ңм§Җ лҶ’м•„м§Ҳ мҲҳ мһҲлҠ”лҚ°, кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ к°ҷмқҖ н•©лі‘мҰқмқҙ мқјл°ҳмқём—җ 비н•ҙ мЎ°кё°м—җ мғқкёҙлӢӨ[5]. к·ёлҹ¬лҜҖлЎң FH мЎ°кё° 진лӢЁмқ„ мң„н•ң м Ғк·№м Ғмқё м„ лі„ кІҖмӮ¬к°Җ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ. н•ңкөӯмқё м Җл°ҖлҸ„ м§ҖлӢЁл°ұ мҪңл ҲмҠӨн…ҢлЎӨ(low-density lipoprotein-cholesterol, LDL-C) мҲҳм№ҳ 분нҸ¬лҘј мғқк°Ғн•ҙліҙл©ҙ, м§ҖкёҲ нҶөмҡ©лҗҳлҠ” мЈјмҡ” мҷёкөӯ 진лӢЁкё°мӨҖмқҳ кё°мӨҖм№ҳлҘј мӮ¬мҡ© н• л•Ң л§ҺмқҖ мӮ¬лһҢмқҙ FHлЎң 진лӢЁлҗ кІғмңјлЎң мҳҲмғҒн• мҲҳ мһҲлӢӨ. к·ёлҹ¬лӮҳ н•ңкөӯмқё нҷҳмһҗм—җ лҢҖн•ң мһҗлЈҢлҠ” м Ғкі мқҳлЈҢкі„ мў…мӮ¬мһҗл“ӨлҸ„ FHм—җ лҢҖн•ҙ мһҳ м•Ңм§Җ лӘ»н•ҳл©°, мқјл°ҳмқёмқҳ мқҙн•ҙлҸ„лҠ” к·№нһҲ лӮ®лӢӨ. к·ёлһҳм„ң ліёкі лҠ” нҳ„мһ¬ н•ңкөӯмқҳ FH мһҗлЈҢлҘј мҶҢк°ңн•ҳм—¬ кҙҖмӢ¬мқ„ к°–кІҢ н•ҳкі , мқҙ м§Ҳнҷҳмқҳ мЎ°кё° 진лӢЁ мҙү진м—җ лҸ„мӣҖмқҙ лҗҳкІҢ н•ҳлҠ” кІғмқҙ лӘ©м ҒмқҙлӢӨ. лҳҗн•ң м „л¬ёк°Җ н•©мқҳм•Ҳмқ„ м •лҰ¬н•ҳм—¬ FH нҷҳмһҗм—җ лҢҖн•ң м Ғм Ҳн•ң мөңмӢ м№ҳлЈҢм—җ лҢҖн•ҙ м•ҲлӮҙн•ҳкі мһҗ н•ңлӢӨ.
ліё лЎ
FHмқҳ мһ„мғҒм Ғ мң м „н•ҷм Ғ нҠ№м„ұ
м „нҳ•м Ғмқё мӢ мІҙ мҶҢкІ¬мқҖ кұҙмқҳ нҷ©мғүмў…(Fig. 1A)кіј мЎ°кё° л°ңмғқ к°Ғл§үнҷҳмқҙлӢӨ. к·ёл Үм§Җл§Ң мқҙ мҶҢкІ¬мқҳ лҜјк°җлҸ„лҠ” лӮ®кі нҷҳмһҗ лӢӨмҲҳм—җм„ң лӮҳнғҖлӮҳм§Җ м•ҠлҠ”лӢӨ. лҳҗн•ң л°ңлӘ© мёЎл©ҙ Xл Ҳмқҙ мҙ¬мҳҒм—җм„ң м•„нӮ¬л ҲмҠӨкұҙмқҙ л‘җкәјмҡҙ кІғмқҙ нҷ•мқёлҗҳкё°лҸ„ н•ңлӢӨ(Fig. 1B). лӢӨлҰ¬ н•ҳл¶Җ лјҲмҷҖ л°ңл°”лӢҘмқ„ 90лҸ„к°Җ лҗҳкІҢ н•ҳкі , мҙ¬мҳҒ кұ°лҰ¬лҠ” 120 cm м •лҸ„лЎң н•ҳл©°, 50 kVмҷҖ 5.0 mAs мЎ°кұҙ н•ҳм—җ мҙ¬мҳҒн•ңлӢӨ[6]. м•„нӮ¬л ҲмҠӨкұҙ л‘җк»ҳмҷҖ нҸӯмқҖ мҙҲмқҢнҢҢлЎң кІҖмӮ¬н•ҳлҠ” кІғлҸ„ к°ҖлҠҘн•ҳлӢӨ(Fig. 1C). л°ңлӘ©мқ„ 90лҸ„лЎң н•ҳкі , мҳҒмғҒмқҖ нҡЎлӢЁкіј мў…лӢЁмңјлЎң м–»лҠ”лӢӨ. н•ңкөӯ FH л“ұлЎқмӮ¬м—… 2020м—җм„ңлҠ” мһ„мғҒ진лӢЁлҗң FH нҷҳмһҗмқҳ мҙқмҪңл ҲмҠӨн…ҢлЎӨкіј LDL-C мӨ‘к°„к°’мқҙ к°Ғк°Ғ 306кіј 221 mg/dLмҳҖмңјл©°, кұҙ нҷ©мғүмў…, кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ мң лі‘лҘ мқҙ 20%, 19%мҳҖлӢӨ. л“ұлЎқлҗң нҷҳмһҗ мӨ‘ 60%, 36%, 3%м—җм„ң мӨ‘мҰқ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқ, мЎ°кё° л°ңлі‘ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ, кұҙ нҷ©мғүмў…мқҳ к°ҖмЎұл Ҙмқҙ к°Ғк°Ғ мһҲм—ҲлӢӨ[7].
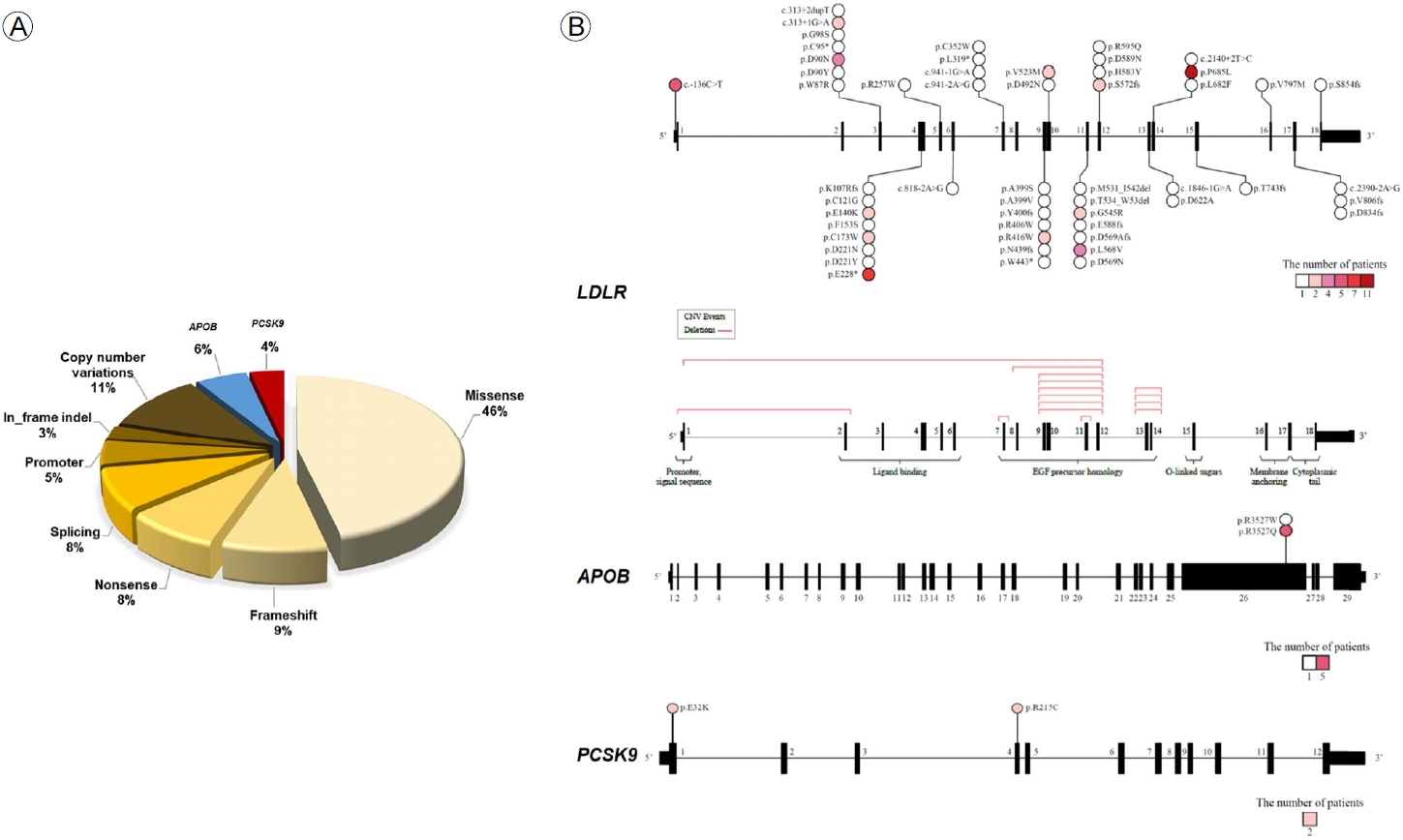
мқҙ м—°кө¬м—җм„ң нҷҳмһҗ 296лӘ… мӨ‘ 104лӘ…(35.1%)м—җм„ң лі‘мқём„ұ ліҖмқҙк°Җ мһҲм—ҲлӢӨ. кіјмҳӨ ліҖмқҙ(missense variant), мң м „мһҗліөм ңмҲҳ ліҖмқҙ(copy number variation), нӢҖмқҙлҸҷ ліҖмқҙ(frameshift variant) мҲңм„ңлҢҖлЎң нқ”н•ҳмҳҖлӢӨ(Fig. 2A). LDLR ліҖмқҙ мӨ‘ м ңмқј нқ”н•ң мң„м№ҳлҠ” p.P685Lмқҙм—ҲлӢӨ(Fig. 2B) [7].
FHмқҳ мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„
м№ҳлЈҢн•ҳм§Җ м•Ҡмңјл©ҙ мқҙнҳ•м ‘н•© FH (heterozygous FH, HeFH) нҷҳмһҗлҠ” 55м„ё(лӮЁмһҗ) нҳ№мқҖ 60м„ё(м—¬мһҗ) мқҙм „м—җ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ мң„н—ҳлҸ„к°Җ мөңлҢҖ 10л°°к№Ңм§Җ лҶ’мқ„ мҲҳ мһҲмңјл©°, нҠ№нһҲ definiteлӮҳ probableнҳ•м—җм„ң к·ёлҹ¬н•ҳлӢӨ[5]. н•ңкөӯм§Җм§ҲлҸҷл§ҘкІҪнҷ”н•ҷнҡҢм—җм„ң м§Җмӣҗлҗң н•ң м—°кө¬м—җм„ң 230л§Ң лӘ… м •лҸ„мқҳ көӯлӮҙ мҪ”нҳёнҠёлҘј 분м„қн•ҳмҳҖлҠ”лҚ°, мӨ‘к°„к°’ 6.1л…„к°„ 추м Ғн•ң кІ°кіј LDL-C 190-224, 225-259, вүҘ 260 mg/dLмқё нҷҳмһҗкө°м—җм„ң мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„(мӢ¬к·јкІҪмғүмҰқ, кҙҖмғҒлҸҷл§Ҙ мһ¬к°ңнҶө, н—ҲнҳҲм„ұ лҮҢмЎёмӨ‘)к°Җ LDL-C < 160 mg/dLмқё кө°м—җ 비н•ҙ мөңлҢҖ 2.4л°° лҶ’м•ҳлӢӨ. LDL-C вүҘ 190 mg/dLмқё нҷҳмһҗм—җм„ң мҙқмӮ¬л§қлҘ мқҖ мөңлҢҖ 2.3л°°мҳҖлӢӨ[8].
FH нҷҳмһҗм—җм„ң мһҳ м•Ңл Ө진 кі„мӮ°мӢқмқ„ нҶөн•ҙ мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„лҘј мӮ°м¶ңн•ҳлҠ” кІғмқҖ л¶Җм Ғм Ҳн•ҳлӢӨ. FHм—җм„ңлҠ” LDL-C мҲҳм№ҳк°Җ м–ҙлҰҙл•Ңл¶Җн„° лҶ’м•„м§Җкё° л•Ңл¬ём—җ, мқҙлҹ° кі„мӮ°мӢқмқҙ мң„н—ҳлҸ„лҘј кіјмҶҢнҸүк°Җ н• мҲҳ мһҲкё° л•Ңл¬ёмқҙлӢӨ[9]. л”°лқјм„ң м§Җм§Ҳк°•н•ҳ м№ҳлЈҢлҘј мң„н•ң лҜёкөӯ м§Җм№Ём—җм„ңлҠ” LDL-C вүҘ 190 mg/dLмқё мӮ¬лһҢл“Өмқ„ мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„к°Җ м•Ҫл¬ј м№ҳлЈҢлҘј л°ӣмқ„ м •лҸ„лЎң лҶ’мқҖ, мҶҢмң„ мҠӨнғҖнӢҙ м№ҳлЈҢ мқҙл“қкө°мңјлЎң 분лҘҳн•ңлӢӨ[10]. 2019л…„ мң лҹҪм§Җм№ЁмқҖ FH нҷҳмһҗлҘј лӢӨлҘё мң„н—ҳ мҡ”мқё мң л¬ҙм—җ л”°лқј мҙҲкі мң„н—ҳкө° нҳ№мқҖ кі мң„н—ҳкө°мңјлЎң 분лҘҳн•ҳмҳҖлӢӨ[5]. м—¬кё°м„ң лӢӨлҘё мң„н—ҳмҡ”мқёмқҖ м „нҶөм Ғ мҡ”мқёмқ„ мқјм»«лҠ”лҚ° лӮҳмқҙ, лӮЁмһҗ, кі нҳҲм••, нқЎм—°, LDL-C, мЈҪмғҒлҸҷл§ҘкІҪнҷ”м„ұ мӢ¬нҳҲкҙҖ м§Ҳнҷҳ, мІҙм§Ҳлҹүм§ҖмҲҳлҘј нҸ¬н•Ён•ңлӢӨ[11]. н•ңкөӯм§Җм§ҲлҸҷл§ҘкІҪнҷ”н•ҷнҡҢм—җм„ң м§Җмӣҗн•ң н•ңкөӯ л“ұлЎқмӮ¬м—…м—җм„ңлҠ” FH нҷҳмһҗм—җм„ң кі нҳҲм••кіј лӮ®мқҖ кі л°ҖлҸ„ м§ҖлӢЁл°ұ мҪңл ҲмҠӨн…ҢлЎӨ(high-density lipoprotein-cholesterol, HDL-C) мҲҳм№ҳк°Җ мһ„мғҒ кІ°кіјм—җ лҢҖн•ң мҳҲмёЎ мҡ”мқёмқҙм—ҲлӢӨ[12].
FHмқҳ 진лӢЁ
мһ„мғҒ진лӢЁ
м„ёкі„м ҒмңјлЎң Dutch Lipid Clinic Network 진лӢЁкё°мӨҖмқҙ м ңмқј л§Һмқҙ мӮ¬мҡ©лҗҳл©°(Table 1) [5], Simon Boome кё°мӨҖлҸ„ нқ”нһҲ м“°мқёлӢӨ(Table 2) [13]. м§ҖкёҲмңјлЎңм„ңлҠ” н•ңкөӯмқём—җм„ң мқҙ л‘җ к°Җм§Җ мӨ‘ н•ҳлӮҳлҘј м“°лҠ” кІғмқҙ м ңмқј н•©лҰ¬м Ғмқҙлқјкі нҢҗлӢЁлҗңлӢӨ. л‘җ к°Җм§Җ кё°мӨҖ лӘЁл‘җ LDL-C мҲҳм№ҳ, кұҙ нҷ©мғүмў… к°ҷмқҖ мӢ мІҙ мҶҢкІ¬, мЎ°кё° л°ңлі‘ кҙҖмғҒлҸҷл§Ҙ м§ҲнҷҳмқҙлӮҳ мӨ‘мҰқ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқ к°ҖмЎұл Ҙ, DNA лҸҢм—°ліҖмқҙлҘј нҸ¬н•Ён•ңлӢӨ. мқҙл°–м—җ лӘҮ лӮҳлқјм—җ лҸ…мһҗм Ғмқё FH мһ„мғҒ진лӢЁкё°мӨҖмқҙ мһҲлӢӨ. мәҗлӮҳлӢӨмҷҖ мқјліём—җм„ңлҠ” LDL-C мҲҳм№ҳ, мһ„мғҒ мҶҢкІ¬, к°ҖмЎұл Ҙмқ„ нҸ¬н•Ён•ң лӘЁлҚёмқ„ 분м„қн•ҳкі лҜјк°җлҸ„мҷҖ нҠ№мқҙлҸ„к°Җ мөңм Ғмқё кё°мӨҖмқ„ л§Ңл“Өм—ҲлӢӨ[14,15]. н•ңкөӯ FH л“ұлЎқмӮ¬м—… 2020м—җм„ң DutchлӮҳ Simon Broome кё°мӨҖмңјлЎң definiteлӮҳ probable нҳ•мңјлЎң 분лҘҳлҗң нҷҳмһҗм—җм„ңлҠ” лі‘мқём„ұ ліҖмқҙ ліҙмң мһҗк°Җ 50-64%мҳҖлӢӨ[7]. лӢ№м—°н•ң м–ҳкё°м§Җл§Ң, мӮ¬мҡ©н•ҳлҠ” кё°мӨҖм—җ л”°лқј лҜјк°җлҸ„мҷҖ нҠ№мқҙлҸ„лҠ” м—ӯмғҒкҙҖ кҙҖкі„к°Җ мһҲлӢӨ. к·ёлҹ¬лҜҖлЎң definite нҳ•мқ„ мң„н•ң 진лӢЁкё°мӨҖмқ„ мӮ¬мҡ©н•ҳл©ҙ нҷҳмһҗлҘј л°ңкөҙн• л•Ңм—җлҠ” нҡЁкіјм Ғмқҙм§Җ м•Ҡмңјл©°, м—°мҮ„ м„ лі„ кІҖмӮ¬лҘј н•ҳлҠ” кІҪмҡ°м—җлҠ” нҠ№мқҙлҸ„к°Җ лҶ’мқҖ кё°мӨҖмқ„ м“°лҠ” кІғмқҙ м Ғм Ҳн• кІғмқҙлӢӨ[16]. 진лӢЁ лӘ©м Ғм—җ л”°лқј 진лӢЁкё°мӨҖмқ„ кі лҘҙлҠ” кІғмқҙ лҸ„мӣҖмқҙ лҗңлӢӨлҠ” мқҳлҜёмқҙлӢӨ.
м§ҖкёҲ FH 진лӢЁкё°мӨҖмқ„ лҸ…мһҗм ҒмңјлЎң л§Ңл“Ө л§Ңн•ң н•ңкөӯ мһҗлЈҢлҠ” 충분м№ҳ м•ҠлӢӨ. к·ёлҹ¬лӮҳ н•ңкөӯ FH л“ұлЎқмӮ¬м—… 2020м—җм„ңлҠ” мқјл°ҳмқёкіј FH нҷҳмһҗм—җм„ң LDL-C мҲҳм№ҳ 분нҸ¬лҘј 분м„қн•ҳмҳҖмңјл©°, мҙқмҪңл ҲмҠӨн…ҢлЎӨ 250 mg/dLмҷҖ LDL-C 177 mg/dLлҘј кІҪкі„к°’мңјлЎң м ңмӢңн•ҳмҳҖлӢӨ(Fig. 3A). н•ңнҺё, лі‘мқём„ұ ліҖмқҙ ліҙмң м—җ лҢҖн•ң кІҪкі„к°’мқҖ мҙқмҪңл ҲмҠӨн…ҢлЎӨкіј LDL-C мҲҳм№ҳ 325 mg/dLмҷҖ 225 mg/dLлЎң лӮҳнғҖлӮ¬лӢӨ(Fig. 3B) [7]. л”°лқјм„ң н•ңкөӯмқём—җм„ң FH м„ лі„ кІҖмӮ¬лӮҳ к°ҖмЎұ лӮҙ м—°мҮ„ м„ лі„ кІҖмӮ¬лҘј н• л•Ң мқҙ мҲҳм№ҳл“Өмқ„ м°ёкі н• мҲҳ мһҲмқ„ кІғмқҙлӢӨ.
мң м „ 진лӢЁ
FHк°Җ мң м „ м§Ҳнҷҳмқҙкё°лҠ” н•ҳм§Җл§Ң мң м „ кІҖмӮ¬лҠ” FHк°Җ мқҳмӢ¬лҗҳлҠ” мӮ¬лһҢ мӨ‘ мқјл¶Җм—җм„ңл§Ң н•ҳкі мһҲлӢӨ. мң м „ кІҖмӮ¬лҠ” 1) нҷ•мӢӨн•ң 진лӢЁ, 2) м„ёл°Җн•ң мң„н—ҳлҸ„ нҸүк°ҖмҷҖ мқҙм—җ л”°лҘё м§Җм§Ҳк°•н•ҳ м№ҳлЈҢ мҙү진, 3) м—°мҮ„ м„ лі„ кІҖмӮ¬ нҡЁмңЁнҷ”[17]м—җ лҸ„мӣҖмқҙ лҗңлӢӨ. мөңк·ј м—°кө¬м—җ л”°лҘҙл©ҙ мӢ мІҙ мҶҢкІ¬мқҙ 비мҠ·н•ң кІҪмҡ°, лі‘мқём„ұ ліҖмқҙк°Җ мһҲлҠ” нҷҳмһҗк°Җ м—ҶлҠ” нҷҳмһҗм—җ 비н•ҙ мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„к°Җ 3л°° лҶ’лӢӨкі н•ңлӢӨ[18]. м–ёкёүн•ҳмҳҖл“Ҝмқҙ, мһ„мғҒ진лӢЁ мӢң 진лӢЁнҳ•(мҳҲ, definite, probable, possible) м—җ л”°лқј лі‘мқём„ұ ліҖмқҙ ліҙмң 비мңЁмқҖ лӢӨлҘј мҲҳ мһҲлӢӨ. FHм—җ л§һлҠ” н‘ңнҳ„нҳ•кіј к°ҖмЎұл Ҙмқҙ мһҲлӢӨл©ҙ, мң м „ кІҖмӮ¬м—җм„ң лі‘мқём„ұ ліҖмқҙк°Җ лӮҳмҳӨм§Җ м•Ҡм•„лҸ„ FH 진лӢЁмқ„ л°°м ңн• мҲҳ м—ҶлӢӨ. мқҙлҹ° мӮ¬лЎҖл“ӨмқҖ лӢӨмң м „мһҗм„ұ(polygenic)мқҙкұ°лӮҳ, кё°мҲ м Ғмқё л¬ём ңкұ°лӮҳ, LDLRAP1 мң м „мһҗмқҳ мғҒм—јмғүмІҙ м—ҙм„ұмқё лі‘мқём„ұ ліҖмқҙкұ°лӮҳ, мғҲлЎңмҡҙ мң м „мһҗ ліҖмқҙм—җ кё°мқён•ң кІҪмҡ°мқј мҲҳ мһҲлӢӨ.
FH мң м „ кІҖмӮ¬м—җм„ң н‘ңм Ғ мң м „мһҗлҠ” LDLR, APOB, PCSK9мқҙлӢӨ. 비мҡ©мқҙ м җм җ лӮ®м•„м§җм—җ л”°лқј мөңк·ј л§ҺмқҖ кё°кҙҖм—җм„ң м°Ём„ёлҢҖ м—јкё°м„ңм—ҙ 분м„қ(next generation sequencing)м—җ кІ°мҶҗ/мӨ‘ліө(deletion/duplication) 분м„қмқ„ лҚ”н•ҙ мқҙ м„ё к°Җм§Җ мң м „мһҗлҘј кІҖмӮ¬н•ңлӢӨ. мҷёкөӯ мһҗлЈҢмҷҖ 비мҠ·н•ҳкІҢ н•ңкөӯ нҷҳмһҗм—җм„ңлҸ„ лі‘мқём„ұ ліҖмқҙ ліҙмң мһҗ мӨ‘ 10% м •лҸ„м—җм„ң мң м „мһҗліөм ңмҲҳліҖмқҙ(copy number variation)к°Җ л°ңкІ¬лҗҳлҠ”лҚ°, мқҙ мў…лҘҳмқҳ ліҖмқҙлҠ” кІ°кіј кІҖмҰқмқ„ мң„н•ҙ лӢӨмӨ‘ кІ°м°°мқҳмЎҙ н”„лЎңлёҢ мҰқнҸӯ(multiplex ligation-dependent probe amplification)мқҙлӮҳ TaqMan л°©лІ•мқҙ н•„мҡ”н•ҳлӢӨ[7]. лі‘мқём„ұ ліҖмқҙлЎң мқҳмӢ¬лҗҳлҠ” ліҖмқҙк°Җ л°ңкІ¬лҗҳлҠ” кІҪмҡ°м—җлҠ” мқҙкІғмқҙ мӣҗмқё ліҖмқҙмқём§Җ мҳ¬л°”лҘҙкІҢ н•ҙм„қн•ҳлҠ” кІғмқҙ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ. LDLR ліҖмқҙм—җ лҢҖн•ң лҚ°мқҙн„°лІ мқҙмҠӨлҠ” м„ёкі„м ҒмңјлЎң лӘҮ к°ңк°Җ мһҲлӢӨ. FH кҙҖл Ё мң м „мһҗмқҳ лі‘мқём„ұмқҖ American College of Medical Genetics and GenomicsмҷҖ Association of Molecular Pathology м§Җм№Ём—җ л”°лқј 분лҘҳн•ңлӢӨ[19]. к·ёл Үм§Җл§Ң л°ңкІ¬лҗҳлҠ” ліҖмқҙ мӨ‘ л§ҺмқҖ мҲҳк°Җ мқҳлҜёк°Җ л¶Ҳнҷ•мӢӨн•ң ліҖмқҙ(variant of uncertain significance)лЎң н•ҙм„қлҗңлӢӨ[20]. мқҙлҹ° кІҪмҡ° ліҖмқҙмқҳ кё°лҠҘм„ұмқҖ к°ҖмЎұ лӮҙ кіөлҸҷ 분лҰ¬(co-segregation)лҘј нҶөн•ҙ нҷ•мқён• мҲҳ мһҲлӢӨ(Fig. 4) [21].
мң м „ кІҖмӮ¬лҘј н• лҢҖмғҒмқҖ нқ”нһҲ м„ұмқём—җм„ң LDL-C вүҘ 190 mg/dLмқҙл©ҙм„ң лӢӨлҘё мқҙм°Ём„ұ мӣҗмқёмқҙ м—Ҷмқ„ л•Ң, мҶҢм•„лӮҳ мІӯмҶҢл…„м—җм„ң LDL-C вүҘ 160 mg/dLмқҙл©ҙм„ң мЎ°кё° л°ңлі‘ кҙҖмғҒлҸҷл§Ҙ м§ҲнҷҳмқҙлӮҳ мӨ‘мҰқ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқ к°ҖмЎұл Ҙмқҙ мһҲлҠ” кІҪмҡ°мқҙлӢӨ. көӯлӮҙм—җм„ң мң м „ кІҖмӮ¬ 비мҡ©мқ„ кұҙк°•ліҙн—ҳм—җм„ң л¶Җ분м ҒмңјлЎң м§Җмӣҗн•ҳкі мһҲмңјлҜҖлЎң, мқҳлЈҢкі„ мў…мӮ¬мһҗл“Өмқҙ FHм—җ лҢҖн•ҙ лҚ” мһҳ м•ҢкІҢ лҗҳл©ҙ нҷҳмһҗм—җ лҢҖн•ң мһ„мғҒм Ғ, мң м „н•ҷм Ғ 진лӢЁмңЁмқҙ мҳ¬лқјк°Ҳ л“Ҝн•ҳлӢӨ.
к°җлі„ 진лӢЁ
лӢӨлҘё мқҙм°Ём„ұ, мқјм°Ём„ұ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқмқҙ л°°м ңлҗҳм–ҙм•ј н•ңлӢӨ. мқҙм°Ём„ұ мӣҗмқём—җлҠ” к°‘мғҒм„ кё°лҠҘ м Җн•ҳмҰқ, мӢ мҰқнӣ„кө°, лӢҙмҰҷ м •мІҙ, кёүм„ұ к°„н—җм„ұ нҸ¬лҘҙн”јлҰ°нҳҲмҰқ, м•Ҫм ң(мҳҲ, thiazide, cyclosporine) л“ұмқҙ мһҲлӢӨ[22]. лӢӨлҘё мқјм°Ём„ұ мӣҗмқёмңјлЎңлҠ” мӢңнҶ мҠӨн…ҢлЎӨнҳҲмҰқ, к°ҖмЎұм„ұ ліөн•© кі м§ҖнҳҲмҰқ л“ұмқҙ мһҲлӢӨ. мӢңнҶ мҠӨн…ҢлЎӨнҳҲмҰқмқҖ нқ¬к·Җ мң м „ м§ҲнҷҳмңјлЎң ABCG5/8мқҳ кё°лҠҘмғҒмӢӨ лҸҢм—°ліҖмқҙм—җ кё°мқён•ҳлҠ”лҚ°, нҳҲмӨ‘ мӢқл¬јм„ұ мҠӨн…ҢлЎӨ лҶҚлҸ„к°Җ лҶ’лӢӨ. 20л§Ң лӘ… мӨ‘ н•ң лӘ… кјҙлЎң л°ңмғқн•ңлӢӨ. LDL-C мҲҳм№ҳлҠ” лӢӨм–‘н•ҳм§Җл§Ң мҶҢм•„ л“ұ мқјл¶Җ нҷҳмһҗм—җм„ңлҠ” л§Өмҡ° лҶ’мңјл©°, мӢқмӮ¬мқҳ мҳҒн–Ҙмқ„ л°ӣмқ„ мҲҳ мһҲлӢӨ. нҷ•м§„мқҖ лі‘мқём„ұ лҸҢм—°ліҖмқҙк°Җ нҷ•мқёлҗҳм–ҙм•ј н•ҳлҠ”лҚ°, мӢңнҶ мҠӨн…ҢлЎӨ мёЎм •кіј мң м „ кІҖмӮ¬лҘј н• мҲҳ м—ҶлҠ” кё°кҙҖмқҙ л§ҺмқҖ кІғмқҙ 진лӢЁмқҳ кұёлҰјлҸҢмқҙлӢӨ[23]. к°ҖмЎұм„ұ ліөн•© кі м§ҖнҳҲмҰқ нҷҳмһҗлҠ” мҙҲм Җл°ҖлҸ„ м§ҖлӢЁл°ұ лҶҚлҸ„мҷҖ LDL-C мҲҳм№ҳк°Җ лҶ’кі , HDL-C мҲҳм№ҳк°Җ лӮ®лӢӨ. мң лі‘лҘ мқҖ 100-200лӘ…лӢ№ н•ң лӘ…кјҙмқёлҚ°, мЎ°кё° л°ңлі‘ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳм—җ лҢҖн•ң мЈјмҡ” мӣҗмқё мӨ‘ н•ҳлӮҳлӢӨ. 분мһҗм Ғ л°°кІҪмқҖ л¶Ҳнҷ•мӢӨн•ҳм§Җл§Ң лӢӨмң м „мһҗм„ұ мӣҗмқёмңјлЎң м„ӨлӘ…лҗҳкё°лҸ„ н•ҳл©°, мқҙм°Ём„ұ мҡ”мқёл“ӨлҸ„ н‘ңнҳ„нҳ•м—җ мҳҒн–Ҙмқ„ мӨ„ мҲҳ мһҲлӢӨ. нҷҳмһҗл“ӨмқҖ м „нҳ•м ҒмңјлЎң мҙқ мҪңл ҲмҠӨн…ҢлЎӨ мҲҳм№ҳк°Җ 200-400 mg/dL, мӨ‘м„ұм§Җл°© 200-600 mg/dL, HDL-C < 40-50 mg/dLмқҙл©°, к°ҖмЎұл Ҙмқҙ мһҲлӢӨ. мқҙ м§Ҳнҷҳм—җ лҢҖн•ҙ м§ҖкёҲк№Ңм§Җ н‘ңмӨҖнҷ”лҗң м •мқҳк°Җ м—Ҷм§Җл§Ң, APOB (вүҘ 120 mg/dL)мҷҖ мӨ‘м„ұм§Җл°©(вүҘ 133 mg/dL)мқҙ лҸҷмӢңм—җ лҶ’мқҖ кІғмқҙ м ңмқј нқ”н•ң лӢЁм„ңмқҙлӢӨ[24].
FHмқҳ м„ лі„ кІҖмӮ¬
м—¬лҹ¬ лӮҳлқјм—җм„ң FH мң лі‘лҘ мқҖ кё°мЎҙм—җ ліҙкі лҗң мһҗлЈҢліҙлӢӨ лҶ’мқ„ к°ҖлҠҘм„ұмқҙ мһҲлҠ”лҚ°, мқҙлҠ” FHмқҳ мһ„мғҒм Ғ мӨ‘мҡ”м„ұм—җ лҢҖн•ң мқёмӢқмқҙ л¶ҖмЎұн•ҙм„ң 진лӢЁмқҙ лҚң лҗҳкё° л•Ңл¬ёмқҙлӢӨ. FHм—җ лҢҖн•ң м§Җм№Ёкіј н•©мқҳм•Ҳл“ӨмқҖ мЎ°кё° 진лӢЁмқ„ мң„н•ҙ м„ лі„ кІҖмӮ¬, нҠ№нһҲ м—°мҮ„ м„ лі„ кІҖмӮ¬мқҳ мӨ‘мҡ”м„ұмқ„ м–ёкёүн•ҳкі мһҲлӢӨ[6,17,25,26]. лҳҗн•ң к°Ғ көӯк°Җмқҳ мғҒнҷ©кіј н•©мқҳм•Ҳм—җ л§һкІҢ м„ лі„ кІҖмӮ¬лҘј н•ҳлҠ” кІғмқ„ м ңм•Ҳн•ңлӢӨ. FH нҷҳмһҗл“Өмқҙ мЈҪмғҒлҸҷл§ҘкІҪнҷ”м„ұ мӢ¬нҳҲкҙҖ м§Ҳнҷҳмқҙ мғқкё°кё° м „к№Ңм§ҖлҠ” лҢҖк°ң мҰқмғҒмқҙ м—Ҷкё° л•Ңл¬ём—җ LDL-C мҲҳм№ҳлҘј мёЎм •н•ҳлҠ” кІғмқҖ мЎ°кё° 진лӢЁкіј м№ҳлЈҢлҘј мң„н•ҙ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ. 2018л…„ н•ңкөӯ мқҙмғҒм§Җм§ҲнҳҲмҰқ м§Җм№ЁмқҖ 21м„ё мқҙмғҒ м„ұмқё нҳ№мқҖ лҚ” м–ҙлҰ¬лҚ”лқјлҸ„ лӢӨлҘё мң„н—ҳмҡ”мқё(мҳҲ, мӢ¬нҳҲкҙҖ м§ҲнҷҳмқҙлӮҳ мӨ‘мҰқ мқҙмғҒм§Җм§ҲнҳҲмҰқ к°ҖмЎұл Ҙ)мқҙ мһҲлҠ” кІҪмҡ° 4-6л…„м—җ н•ң лІҲ мқҙмғҒм§Җм§ҲнҳҲмҰқм—җ лҢҖн•ң м„ лі„ кІҖмӮ¬лҘј н• кІғмқ„ к¶Ңкі н•ҳкі мһҲлӢӨ[27].
кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳмқҙ < 55м„ё(лӮЁмһҗ) нҳ№мқҖ < 60м„ё(м—¬мһҗ)м—җ мғқкІјмқ„ л•Ң нҳ№мқҖ LDL-C мҲҳм№ҳк°Җ мӢ¬н•ҳкІҢ лҶ’мқ„ л•Ң(м„ұмқём—җм„ң вүҘ 190 mg/dL нҳ№мқҖ мҶҢм•„м—җм„ң вүҘ 150 mg/dL), нҳ№мқҖ нҷҳмһҗ мһҗмӢ нҳ№мқҖ к°ҖмЎұ, м№ңмІҷ мӨ‘ кұҙ нҷ©мғүмў…мқҙ мһҲмқ„ л•Ң нҳ№мқҖ мЎ°кё° л°ңлі‘ мӢ¬нҳҲкҙҖ м§Ҳнҷҳ к°ҖмЎұл Ҙмқҙ мһҲмқ„ л•Ң FHлҘј мқҳмӢ¬н•ҙм•ј н•ңлӢӨ[5,27]. н”јл¶Җ нҷ©мғүмў…, xanthelasma, мЎ°кё° л°ңмғқ к°Ғл§үнҷҳлҸ„ FH м„ лі„ кІҖмӮ¬лҘј мң„н•ң кі л ӨмӮ¬н•ӯмқҙлӢӨ. к·ёлҹ¬лӮҳ л§ҺмқҖ FH нҷҳмһҗк°Җ мқҙ м§ҲнҷҳмңјлЎң мқҳмӢ¬лҗҳкё° м „м—җ м§Җм§Ҳк°•н•ҳ м•Ҫм ңлҘј мқҙлҜё мӢңмһ‘н•ҳкё° мү¬мҡ°л©°, мқҙ л•Ңл¬ём—җ 진лӢЁмқҙ м§Җм—°лҗҳлҠ” кІҪн–Ҙмқҙ мһҲлӢӨ. к·ёлһҳм„ң FHм—җ л§һлҠ” мһ„мғҒ мҶҢкІ¬кіј к°ҖмЎұл Ҙмқ„ нҷ•мқён• мҲҳ мһҲкІҢ мқҳлЈҢкі„ мў…мӮ¬мһҗ мӮ¬мқҙм—җ FH мқём§ҖлҸ„лҘј к°ңм„ н•ҳлҠ” кІғмқҙ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ.
к°ҖмЎұ лӮҙм—җ мІҳмқҢ 진лӢЁлҗң index нҷҳмһҗмқҳ к°ҖмЎұм—җ лҢҖн•ң м—°мҮ„ м„ лі„ кІҖмӮ¬лҠ” мғҲлЎңмҡҙ FH нҷҳмһҗлҘј мЎ°кё°м—җ 진лӢЁ, м№ҳлЈҢн•ҳкё° мң„н•ң нҡЁмңЁкіј к°Җм„ұ비к°Җ м ңмқј мўӢмқҖ л°©лІ•мңјлЎң мһҳ м•Ңл Өм ё мһҲлӢӨ. м—°мҮ„ м„ лі„ кІҖмӮ¬лҠ” мқјм°Ём Ғ к°ҖмЎұ, м№ңмІҷм—җ лҢҖн•ң м§Җм§Ҳ мҲҳм№ҳмҷҖ мң м „ кІҖмӮ¬лҘј нҸ¬н•Ён•ңлӢӨ. мһҳ кө¬м„ұлҗң м„ лі„ кІҖмӮ¬ н”„лЎңк·ёлһЁмқҙ FHмқҳ мҳҲнӣ„лҘј к°ңм„ н• мҲҳ мһҲмңјлҜҖлЎң л§ҺмқҖ лӮҳлқјм—җм„ң мһҗкөӯм—җ м Ғм Ҳн•ң н”„лЎңк·ёлһЁмқ„ лҸ„мһ…н•ҳкі мһҲлӢӨ. мқјкҙ„(universal) м„ лі„ кІҖмӮ¬лҠ” FH нҷҳмһҗ л°ңкөҙмқ„ мң„н•ң л°©лІ• мӨ‘ н•ҳлӮҳмқёлҚ°, мқјл¶Җ көӯк°Җм—җм„ң мӢңн–үлҗҳкі мһҲлӢӨ[13].
FHмқҳ м№ҳлЈҢ
FHк°Җ 진лӢЁлҗҳл©ҙ м§Җм§Ҳк°•н•ҳ м№ҳлЈҢлҘј лҗҳлҸ„лЎқ л№ЁлҰ¬ мӢңмһ‘н•ҳлҠ” кІғмқҙ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ. лӢӨлҘё мӢ¬нҳҲкҙҖ мң„н—ҳ мҡ”мқёмқҙ мһҲлӢӨл©ҙ лҸҷмӢңм—җ мЎ°м Ҳн•ҳлҠ” кІғлҸ„ н•„мҲҳм ҒмқёлҚ°, мқҙлҠ” FH м№ҳлЈҢ лӘ©м Ғмқҙ мЈҪмғҒлҸҷл§ҘкІҪнҷ”м„ұ мӢ¬нҳҲкҙҖ м§Ҳнҷҳ мҳҲл°©м—җ мһҲкё° л•Ңл¬ёмқҙлӢӨ. мӢқмӮ¬ мЎ°м ҲмқҙлӮҳ мҡҙлҸҷк°ҷмқҖ 비м•Ҫл¬ј м№ҳлЈҢлҠ” мқҙмғҒм§Җм§ҲнҳҲмҰқм—җ лҢҖн•ң лӢӨлҘё м§Җм№Ёкіј лҢҖлҸҷмҶҢмқҙн•ҳлӢӨ[13,27]. к°„лһөнһҲ л§җн•ҳл©ҙ мҙқ м§Җл°©, нҸ¬нҷ”м§Җл°©, нҠёлһңмҠӨм§Җл°©, мҪңл ҲмҠӨн…ҢлЎӨ, нғ„мҲҳнҷ”л¬ј, лӢ№лҘҳ, м•ҢмҪ”мҳ¬ м„ӯм·ЁлҠ” м ңн•ңн•ҳл©°, мқҙл“Өмқҳ мғҒн•ңм„ мқ„ м ңмӢңн•ҳкі мһҲлӢӨ. н•ңнҺё, 섬мң мҶҢк°Җ н’Қл¶Җн•ң мқҢмӢқ, нҶөкіЎ, лӢӨм–‘н•ң кіЎлҘҳ, м•јмұ„, мғқм„ , мӢ м„ н•ң кіјмқјмқ„ м Ғк·№м ҒмңјлЎң лЁ№мқ„ кІғмқ„ к¶Ңкі н•ңлӢӨ. мң мӮ°мҶҢ мҡҙлҸҷ, м Җн•ӯм„ұ мҡҙлҸҷмқ„ к·ңм№ҷм ҒмңјлЎң н•ҳлҠ” кІғмқҙ 추мІңлҗҳлҠ”лҚ°[27], мӨ‘к°„ к°•лҸ„ мң мӮ°мҶҢ мҡҙлҸҷмқ„ мқјмЈјмқјм—җ 4-6нҡҢ, 30분 мқҙмғҒ н•ҳл©°, мқјмЈјмқјм—җ 2нҡҢ мқҙмғҒ м Җн•ӯм„ұ мҡҙлҸҷмқ„ н•ҳлҠ” кІғмқ„ к¶Ңкі н•ҳкі мһҲлӢӨ.
мқјм°Ём Ғмқё м№ҳлЈҢм•Ҫм ңлҠ” мҠӨнғҖнӢҙмқҙл©°, лҢҖк°ң кі к°•лҸ„лЎң нҲ¬м—¬н•ҳкІҢ лҗңлӢӨ. л§ҺмқҖ FH нҷҳмһҗк°Җ мҠӨнғҖнӢҙ лӢЁлҸ…мҡ”лІ•мңјлЎң LDL-C лӘ©н‘ңм№ҳм—җ лҸ„лӢ¬н•ҳм§Җ лӘ»н• мҲҳ мһҲмңјл©°, мқҙм°Ё м•Ҫм ңлЎң м—җм ңнӢ°лҜёлёҢк°Җ 추к°Җлҗ мҲҳ мһҲлӢӨ. кІ¬л”ң мҲҳ мһҲлҠ” мөңлҢҖмҡ©лҹү мҠӨнғҖнӢҙмқ„ нҲ¬м—¬н•ҳкі м—җм ңнӢ°лҜёлёҢк°Җ 추к°Җлҗң л’Өм—җлҸ„ лӘ©н‘ңм№ҳм—җ лҸ„лӢ¬н•ҳм§Җ лӘ»н•ҳл©ҙ, PCSK9 м–өм ңм ңлҘј 추к°Җн• мҲҳ мһҲлӢӨ(Table 3) [5,15,28,29]. н•ңкөӯ м§Җм§ҲлҸҷл§ҘкІҪнҷ”н•ҷнҡҢм—җм„ң м§Җмӣҗн•ң н•ң м—°кө¬м—җ л”°лҘҙл©ҙ н•ңкөӯмқё FH нҷҳмһҗм—җм„ң мөңлҢҖмҡ©лҹү мҠӨнғҖнӢҙ/м—җм ңнӢ°лҜёлёҢ лі‘н•©мҡ”лІ•мқ„ н•ң л’Өм—җ LDL-C < 100 mg/dLлӮҳ LDL-C 50% кІҪк°җмқ„ лӢ¬м„ұн•ң 비мңЁмқҖ лҶ’м§Җ м•Ҡм•ҳлӢӨ[30]. м§Җм§Ҳк°•н•ҳ м№ҳлЈҢм—җ лҢҖн•ң нҡЁкіјк°Җ FH нҷҳмһҗмқҳ мң м „нҳ•мқҳ мҳҒн–Ҙмқ„ л°ӣмқ„ мҲҳ мһҲлӢӨ[31].
мҠӨнғҖнӢҙ: лҢҖл¶Җ분мқҳ көӯм ңм Ғ м§Җм№Ёмқҙ нҳ„мһ¬ FH нҷҳмһҗм—җ лҢҖн•ң мқјм°Ё м•Ҫм ңлЎң кІ¬л”ң мҲҳ мһҲлҠ” мөңлҢҖ мҡ©лҹү мҠӨнғҖнӢҙмқ„ к¶Ңкі н•ңлӢӨ(Table 3) [5,29]. FH нҷҳмһҗм—җм„ң мҠӨнғҖнӢҙм—җ лҢҖн•ң л¬ҙмһ‘мң„ л°°м • мһ„мғҒмӢңн—ҳмқҖ мҲҳн–үлҗң м Ғмқҙ м—ҶлӢӨ. н•ҳм§Җл§Ң л„ӨлҚңлһҖл“ңм—җм„ң мҲҳн–үлҗң мҪ”нҳёнҠё м—°кө¬м—җм„ң мөңмӢ м§Җм№ЁліҙлӢӨ лӮ®мқҖ мҡ©лҹү мҠӨнғҖнӢҙмқ„ мӮ¬мҡ©н•ҳмҳҖлҠ”лҚ°лҸ„ FH нҷҳмһҗм—җм„ң кҙҖмғҒлҸҷл§Ҙм„ұ мӢ¬мһҘ м§Ҳнҷҳ мң„н—ҳлҸ„к°Җ 76% лӮ®лӢӨкі ліҙкі н•ҳмҳҖлӢӨ[32]. лҳҗн•ң л„ӨлҚңлһҖл“ңм—җм„ң мқҙлЈЁм–ҙ진 нӣ„н–Ҙм Ғ м—°кө¬м—җм„ң мӨ‘к°„ к°•лҸ„-кі к°•лҸ„ мҠӨнғҖнӢҙ м№ҳлЈҢ мқҙнӣ„ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ л°ңмғқкіј мӮ¬л§қлҘ мқҙ 50% к°җмҶҢлҗЁмқ„ нҷ•мқён•ҳмҳҖлӢӨ[33]. л©”нғҖ분м„қ м—°кө¬м—җм„ңлҸ„ кі к°•лҸ„ мҠӨнғҖнӢҙ м№ҳлЈҢк°Җ мң мқөн•Ёмқ„ м•Ң мҲҳ мһҲлӢӨ[34].
м—җм ңнӢ°лҜёлёҢ: лҢҖл¶Җ분мқҳ мөңк·ј м§Җм№Ём—җм„ң м—җм ңнӢ°лҜёлёҢлҠ” м§Җм§Ҳ к°•н•ҳ м№ҳлЈҢмқҳ мқҙм°Ё м•Ҫм ңлЎң к¶Ңкі лҗңлӢӨ(Table 3). мҪңл ҲмҠӨн…ҢлЎӨ нқЎмҲҳ м–өм ңм ңлЎңм„ң мӨ‘к°„ к°•лҸ„ мҠӨнғҖнӢҙкіј лі‘н•©н•ҳмҳҖмқ„ л•Ң мӢ¬нҳҲкҙҖ мӮ¬кұҙ л°ңмғқмқ„ кІҪк°җн•ҳлҠ” кІғмқҙ м•Ңл Өм ё мһҲлӢӨ[35]. лӢӨлҘё н•ң м—°кө¬лҠ” мҠӨнғҖнӢҙкіј м—җм ңнӢ°лҜёлёҢ лі‘н•©мҡ”лІ•мқҙ мҠӨнғҖнӢҙ лӢЁлҸ…мҡ”лІ•кіј 비көҗн• л•Ң кІҪнҷ”л°ҳмқ„ нҮҙн–үмӢңнӮЁлӢӨкі ліҙкі н•ҳмҳҖлӢӨ[36]. мҠӨнғҖнӢҙ/м—җм ңнӢ°лҜёлёҢ лі‘н•©мҡ”лІ•мқҖ LDL-C к°•н•ҳнҡЁкіјк°Җ к°•н•ҳл©°(кё°м Җ мҲҳм№ҳлЎңл¶Җн„° вүҘ 50%) 비көҗм Ғ м•Ҳм „н•ң м§Җм§Ҳк°•н•ҳ м№ҳлЈҢмқҙлӢӨ[37].
PCSK9 м–өм ңм ң-лӢЁнҒҙлЎ н•ӯмІҙ: мқҙ м•Ҫм ңлҠ” мӢ¬нҳҲкҙҖ мң„н—ҳлҸ„к°Җ л§Өмҡ° лҶ’мқҖ FH нҷҳмһҗм—җм„ң кІ¬л”ң мҲҳ мһҲлҠ” мөңлҢҖмҡ©лҹү мҠӨнғҖнӢҙ/м—җм ңнӢ°лҜёлёҢлҘј мӮ¬мҡ©н•ң л’Өм—җлҸ„ LDL-C лӘ©н‘ңм№ҳм—җ лҸ„лӢ¬н•ҳм§Җ лӘ»н•ң кІҪмҡ° к¶Ңкі лҗңлӢӨ(Table 3). FOURIER [38]мҷҖ ODYSSEY-OUTCOMES м—°кө¬[39]к°Җ мқҙ кі„м—ҙ м•Ҫм ңмқё evolocumabкіј alirocumabмқҳ мӢ¬нҳҲкҙҖ мқҙл“қмқ„ к°Ғк°Ғ мҰқлӘ…н•ҳмҳҖлҠ”лҚ°, мқҙкІғмқҙ FH нҷҳмһҗм—җм„ң н•ӯ-PCSK9 н•ӯмІҙ мӮ¬мҡ©м—җ лҢҖн•ң кіјн•ҷм Ғ к·јкұ°лҘј л§Ҳл Ён•ҳмҳҖлӢӨ. мқҙл“Ө м•Ҫм ңм—җ мқҳн•ң LDL-C к°•н•ҳ м •лҸ„лҠ” FHк°Җ м—ҶлҠ” нҷҳмһҗм—җм„ңліҙлӢӨ мһ‘м§Җ м•ҠлӢӨ[5]. мқҙ м•Ҫм ңл“ӨмқҖ мҠӨнғҖнӢҙ л¶Җмһ‘мҡ©мқ„ кІӘмқҖ нҷҳмһҗл“Өм—җкІҢлҸ„ кі л ӨлҗңлӢӨ. к·ёл Үм§Җл§Ң лӢӨлҘё кІҪкө¬ м•Ҫм ңліҙлӢӨ 비мӢјлҚ°, л”°лқјм„ң нҠ№м • мң„н—ҳкө°м—җм„ң к°Җм„ұ비лҘј кі л Өн•ҳм—¬ н•ӯ PCSK9 н•ӯмІҙлҘј мӢңмһ‘н• м Ғм Ҳн•ң LDL-CлҘј м •н•ҳлҠ” кІғмқҖ м–ҙл Өмҡ°л©ҙм„ңлҸ„ мӨ‘мҡ”н•ң л¬ём ңмқҙлӢӨ[29,40,41].
лӢҙмҰҷмӮ° кІ°н•©мҲҳм§Җ: мӨ‘мҰқ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқм—җм„ң мқҙ м•Ҫм ң 추к°ҖлҘј кі л Өн• мҲҳ мһҲлӢӨ(Table 3). лӢҙмҰҷмӮ° кІ°н•©мҲҳм§ҖлҠ” LDL-C к°•н•ҳ нҡЁкіјк°Җ мһҲм§Җл§Ң, мһ„мғҒ кІҪкіјлҘј ліё м—°кө¬к°Җ м—Ҷкё° л•Ңл¬ём—җ мӮ¬мҡ© к¶Ңкі лҠ” 비көҗм Ғ м ңн•ңм ҒмқҙлӢӨ.
кё°нғҖ м№ҳлЈҢмҷҖ мғҲлЎңмҡҙ м№ҳлЈҢ: mipomersenмқҖ мҳ¬лҰ¬кі лүҙнҒҙл ҲмҳӨнӢ°л“ң мң лҸ„мІҙмқҙл©°, APOB mRNAмҷҖ кІ°н•©н•ҳм—¬ лӘЁл“ APOB н•Ёмң м§ҖлӢЁл°ұ мғқмӮ°мқ„ м–өм ңн•ңлӢӨ. мқҙ м•Ҫм ңмқҳ нҷңм„ұмқҖ LDLR л°ңнҳ„м—җ 비мқҳмЎҙм ҒмқёлҚ°, нҠ№нһҲ лҸҷнҳ•м ‘н•© FH (homozygous FH, HoFH) нҷҳмһҗлҘј мң„н•ң ліҙмЎ° м•Ҫм ңлЎңм„ң к°ңл°ңлҗҳм—ҲлӢӨ. мқҙ нҷҳмһҗм—җм„ң mipomersenмқҖ LDL-C мҲҳм№ҳлҘј 21% к°•н•ҳмӢңмј°лӢӨ. л¶Җмһ‘мҡ©мқҖ мЈјмӮ¬ л¶Җмң„ л°ҳмқ‘, к°„ нҡЁмҶҢмҲҳм№ҳ мғҒмҠ№, к°„ м§Җл°©лҹү мҰқк°Җ л“ұмқҙлӢӨ[42]. нҳ„мһ¬ мқҙ м•Ҫм ңлҠ” көӯлӮҙм—җм„ң мң нҶөлҗҳм§Җ м•ҠлҠ”лӢӨ. LomitapideлҠ” microsomal triglyceride transfer protein м–өм ңм ңмқҙл©°, кіЁм§ҖмІҙм—җм„ң APOB н•Ёмң м§ҖлӢЁл°ұмқҳ мЎ°лҰҪкіј 분비лҘј мӨ„мқёлӢӨ. мқҙ м•Ҫм ңлҠ” HoFH нҷҳмһҗм—җм„ң LDL-C мҲҳм№ҳлҘј 38-50% к°•н•ҳмӢңнӮЁлӢӨ[43]. Lomitapide лҳҗн•ң LDLRм—җ 비мқҳмЎҙм ҒмңјлЎң LDL-C мҲҳм№ҳлҘј к°•н•ҳн•ҳлҠ”лҚ°, к°„м—җ мӨ‘м„ұм§Җл°© 축м Ғ, м§Җл°©к°„м—ј hepatosteatosis), к°„ нҡЁмҶҢ мҲҳм№ҳ мғҒмҠ№мқ„ мҙҲлһҳн• мҲҳ мһҲлӢӨ. мқҙ м•Ҫм ңлҸ„ көӯлӮҙм—җм„ң мң нҶөлҗҳм§Җ м•ҠлҠ”лӢӨ. InclisiranмқҖ н•©м„ұ small interfering RNAмқҙл©°, PCSK9 н•©м„ұмқ„ м–өм ңн•ҳкі LDL-C мҲҳм№ҳлҘј лӮ®м¶”лҠ”лҚ° л§Өмҡ° нҡЁкіјм ҒмқҙлӢӨ. HeFH нҷҳмһҗм—җм„ң мҲҳн–үн•ң мһ„мғҒмӢңн—ҳм—җм„ң мң„м•Ҫм—җ 비н•ҙ LDL-C мҲҳм№ҳлҘј 48% к°•н•ҳмӢңмј°мңјл©°, л¶Җмһ‘мҡ© л№ҲлҸ„лҠ” 비мҠ·н•ҳмҳҖлӢӨ[44]. нҲ¬м—¬ к°„кІ©мқҙ кёём–ҙм„ң нҷҳмһҗ мҲңмқ‘лҸ„к°Җ мўӢмқ„ кІғмңјлЎң кё°лҢҖлҗңлӢӨ. мқҙ м•Ҫм ңлҠ” мӢ¬нҳҲкҙҖ м№ҳлӘ…лҘ кіј мқҙнҷҳмңЁм—җ лҢҖн•ң лҢҖк·ңлӘЁ мһ„мғҒмӢңн—ҳмқҙ 진н–ү мӨ‘мқҙл©°, 2021л…„м—җ лҜёкөӯм—җм„ң мҠ№мқёлҗҳм—ҲмңјлӮҳ м•„м§Ғ көӯлӮҙм—җлҠ” лҸ„мһ…лҗҳм§Җ м•Ҡм•ҳлӢӨ. Angiopoietin-like 3(ANGPTL3)лҠ” lipoprotein lipaseмҷҖ endothelial lipaseлҘј м–өм ңн•ҳлҠ” лӢЁл°ұм§ҲмқҙлӢӨ. EvinacumabмқҖ ANGPTL3м—җ лҢҖн•ң н•ӯмІҙмқҙл©°, HeFH [45]мҷҖ HoFH нҷҳмһҗм—җм„ң[46] к°Ғк°Ғ мҲҳн–үлҗң 2мғҒ, 3мғҒ мһ„мғҒмӢңн—ҳм—җм„ң LDL-Cкіј мӨ‘м„ұм§Җл°© мҲҳм№ҳлҘј мөңлҢҖ 56% к°•н•ҳмӢңмј°лӢӨ.
м§ҖлӢЁл°ұ нҳҲмһҘкөҗнҷҳмҲ мқҖ нҳҲм•ЎмңјлЎңл¶Җн„° м§ҖлӢЁл°ұмқ„ м ңкұ°н•ҳлҠ” л°©лІ•мңјлЎңм„ң, м•Ҫл¬ј м№ҳлЈҢм—җ лҢҖн•ң м§Җм§Ҳк°•н•ҳ л°ҳмқ‘мқҙ 충분м№ҳ м•Ҡмқ„ л•Ң м“°кё°лҸ„ н•ңлӢӨ. мқҙ л°©лІ•мқҖ HoFHлӮҳ мӨ‘мҰқ HeFH нҷҳмһҗм—җм„ң к°ҖлҒ” мӮ¬мҡ©лҗңлӢӨ. мқҙ л°©лІ•мқҖ LDL-C мҲҳм№ҳлҘј 50-70% к°•н•ҳмӢңнӮЁлӢӨ. к·ёл Үм§Җл§Ң м№ЁмҠөм Ғмқё л°©лІ•мқҙкё° л•Ңл¬ём—җ нҷҳмһҗмқҳ мӮ¶мқҳ м§Ҳм—җ л¶Җм •м Ғ мҳҒн–Ҙмқҙ мһҲмқ„ мҲҳ мһҲлӢӨ[5].
LDL-C лӘ©н‘ңм№ҳ: мқҙмғҒм Ғмқё LDL-C лӘ©н‘ңм№ҳлҠ” мЈҪмғҒлҸҷл§ҘкІҪнҷ”м„ұ мӢ¬нҳҲкҙҖ м§ҲнҷҳмқҙлӮҳ мЈјмҡ” мң„н—ҳмҡ”мқёмқҙ мһҲмқ„ л•Ң кё°м Җм№ҳ лҢҖ비 50% кІҪк°җм—җ лҚ”н•ҳм—¬ < 55 mg/dL, л‘җ к°Җм§Җ лӘЁл‘җ м—Ҷмқ„ л•Ң < 70 mg/dLмқҙлӢӨ[5]. н•ҳм§Җл§Ң 3к°Җм§Җ м§Җм§Ҳк°•н•ҳ м•Ҫм ңлҘј лі‘н•©н•ҳлҚ”лқјлҸ„ л§ҺмқҖ FH нҷҳмһҗм—җм„ң мқҙ лӘ©н‘ңм№ҳм—җ лҸ„лӢ¬н•ҳлҠ” кІғмқҙ м–ҙл Өмҡё мҲҳ мһҲкё° л•Ңл¬ём—җ[47], м „мһҗм—җм„ң LDL-C 50% к°•н•ҳм—җ лҚ”н•ҳм—¬ < 70 mg/dL, нӣ„мһҗм—җм„ң < 100 mg/dLк°Җ л§ҺмқҖ м§Җм№Ём—җм„ң нҳ„мӢӨм Ғмқё м°Ём„ мұ…мңјлЎң кұ°лЎ лҗңлӢӨ[6,28,48]. н•ңкөӯм§Җм§ҲлҸҷл§ҘкІҪнҷ”н•ҷнҡҢм—җм„ң м§Җмӣҗн•ң н•ң м—°кө¬м—җм„ң мӨ‘мҰқ кі мҪңл ҲмҠӨн…ҢлЎӨнҳҲмҰқ нҷҳмһҗлҘј 분м„қн•ң кІ°кіј, мӢ¬нҳҲкҙҖ м§Ҳнҷҳмқҙ м—ҶлҠ” нҷҳмһҗм—җм„ң мҠӨнғҖнӢҙ м№ҳлЈҢ нӣ„ LDL-C < 100 mg/dLм—җ лҸ„лӢ¬н•ң нҷҳмһҗм—җм„ң лҸ„лӢ¬н•ҳм§Җ м•ҠмқҖ нҷҳмһҗліҙлӢӨ мӢ¬нҳҲкҙҖ мӮ¬кұҙ л°ңмғқмқҙ м ҒмқҖ кІғмқ„ м•Ң мҲҳ мһҲм—ҲлӢӨ[8].
HoFH
HoFHлҠ” нқ¬к·Җн•ҳм§Җл§Ң м№ҳлӘ…м Ғмқё м§ҲнҷҳмқҙлӢӨ. мң лі‘лҘ мқҖ 100л§ҢлӘ…лӢ№ 1лӘ… м •лҸ„м§Җл§Ң, мөңк·ј м—°кө¬м—җм„ңлҠ” 16-30л§Ң лӘ…лӢ№ 1лӘ…мқҙлқјкі ліҙкі лҗҳкё°лҸ„ н•ңлӢӨ[9]. HeFHліҙлӢӨ HoFH нҷҳмһҗм—җм„ң нҳҲкҙҖмқҙ м§Җм§Ҳм—җ лҢҖн•ҙ л…ём¶ңлҗҳлҠ” м •лҸ„к°Җ лҚ” мӢ¬н•ҳкё° л•Ңл¬ём—җ, 20м„ё мқҙм „м—җ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳмқҙ л°ңмғқн•ҳлҠ” мҳҲлҸ„ л“ңл¬јм§Җ м•ҠлӢӨ.
мһ„мғҒм ҒмңјлЎң кҙ‘лІ”мң„н•ң нҷ©мғүмў…, л§Өмҡ° мЎ°кё°м—җ л°ңлі‘н•ҳкі м§„н–үн•ҳлҠ” мӢ¬нҳҲкҙҖ м§Ҳнҷҳ, м№ҳлЈҢ м „ LDL-C мҲҳм№ҳ > 500 mg/dL нҳ№мқҖ м№ҳлЈҢ нӣ„ LDL-C мҲҳм№ҳ вүҘ 300 mg/dLк°Җ нҠ№м§•мқҙлӢӨ. мғҒлӢ№мҲҳ нҷҳмһҗк°Җ 20м„ё мқҙм „м—җ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳкіј лҢҖлҸҷл§Ҙ нҢҗл§ү нҳ‘м°©мҰқмқҙ мғқкё°кі , 30м„ё мқҙм „м—җ мӮ¬л§қн• мҲҳ мһҲлӢӨ. мҶҢм•„м—җм„ңлҠ” мҙҲкё° мҰқмғҒкіј 징нӣ„лЎңм„ң нҢҗл§үм—җ мҪңл ҲмҠӨн…ҢлЎӨ лӢӨлҹү 축м Ғм—җ кё°мқён•ң лҢҖлҸҷл§Ҙ нҢҗл§үнҳ‘м°©мҰқкіј нҸҗмҮ„л¶Җм „мқҙ лӮҳнғҖлӮ мҲҳ мһҲлӢӨ[25].
진лӢЁ: HoFHм—җ лҢҖн•ҙ м ңмқј мһҳ м•Ңл Ө진 진лӢЁкё°мӨҖмқҖ 2014л…„ мң лҹҪ лҸҷл§ҘкІҪнҷ”н•ҷнҡҢ FH н•©мқҳ нҢЁл„җм—җм„ң лӮҳмҳЁ кІғмқҙлӢӨ[25]. мқҙ кё°мӨҖмқҖ DNA лҸҢм—°ліҖмқҙ, LDL-C мҲҳм№ҳ, мӢ мІҙ мҶҢкІ¬, к°ҖмЎұл Ҙмқ„ нҸ¬н•Ён•ңлӢӨ(Table 4). к·ёлҹ¬лӮҳ мң м „ 진лӢЁкё°мӨҖмқ„ л„Ҳл¬ҙ м—„кІ©н•ҳкІҢ м Ғмҡ©н•ҳл©ҙ м•Ҫл¬ј м№ҳлЈҢм—җ лҢҖн•ң ліҙн—ҳ м Ғмҡ© л“ұм—җ мҳҒн–Ҙмқ„ мӨ„ мҲҳ мһҲлӢӨ. мң м „ 분м„қмқҖ мһ„мғҒ진лӢЁмқ„ нҷ•мқён•ҳкі нҷҳмһҗ к°ҖмЎұм—җ лҢҖн•ң кІҖмӮ¬лҘј мҙү진н•ҳл©°, мһ„мғҒ м–‘мғҒмқҙ HoFHмҷҖ HeFHмқҳ кІҪкі„м„ м—җ мһҲмқ„ л•Ң 진лӢЁмқ„ лҸ•кё° мң„н•ҙ кі л ӨлҗңлӢӨ[25,49].
к°җмӢң: HoFHк°Җ мқҳмӢ¬лҗҳлҠ” нҷҳмһҗлҠ” мў…н•©м Ғмқё мІҳм№ҳлҘј мң„н•ҙ м „л¬ёк°Җм—җкІҢ мқҳлў°н• н•„мҡ”к°Җ мһҲлӢӨ. кҙҖмғҒлҸҷл§Ҙ м§ҲнҷҳмқҙлӮҳ лҢҖлҸҷл§ҘнҢҗл§ү м§Ҳнҷҳм—җ лҢҖн•ң м •кё°м Ғ м„ лі„ кІҖмӮ¬к°Җ к¶Ңкі лҗңлӢӨ. нҷҳмһҗлҠ” 진лӢЁ мӢңм—җ мӢ¬нҳҲкҙҖ нҸүк°ҖлҘј л°ӣкі мқҙнӣ„ л§Өл…„ мӢ¬мһҘмҙҲмқҢнҢҢ, л¶Җн•ҳ кІҖмӮ¬лҘј н•ҳл©°, к°ҖлҠҘн•ң кІҪмҡ° м»ҙн“Ён„°лӢЁмёөкҙҖмғҒлҸҷл§ҘмЎ°мҳҒмҲ мқ„ л§Ө 5л…„л§ҲлӢӨ н•ҳлҠ” кІғмқҙ к¶Ңкі лҗңлӢӨ. мқ‘кёүмғҒнҷ©м—җ лҢҖн•ң көҗмңЎмқҙ н•„мҡ”н•ҳкі , л§Ө 6к°ңмӣ”л§ҲлӢӨ мһ„мғҒм Ғ нҸүк°ҖлҘј н• кІғмқ„ к¶ҢмһҘн•ңлӢӨ.
м№ҳлЈҢ: HoFHлҠ” мҶҢм•„кё°м—җ мқјм°Қ л°ңкІ¬н•ҳлҠ” кІғмқҙ л§Өмҡ° мӨ‘мҡ”н•ҳлӢӨ. HoFHм—җм„ң м№ҳлЈҢ лӘ©м ҒмқҖ м§Җм§Ҳк°•н•ҳ м№ҳлЈҢлҘј мөңлҢҖн•ң мқјм°Қ мӢңмһ‘н•ҳл©°, мҪңл ҲмҠӨн…ҢлЎӨ мҲҳм№ҳлҘј мөңлҢҖн•ң лӮ®м¶”лҠ” кІғмқҙлӢӨ. LDL-C лӘ©н‘ңм№ҳлҠ” м„ұмқё, мҶҢм•„, мЈҪмғҒлҸҷл§ҘкІҪнҷ”м„ұ мӢ¬нҳҲкҙҖ м§Ҳнҷҳ нҷҳмһҗм—җм„ң к°Ғк°Ғ 100, 135, 70 mg/dLмқҙлӢӨ. мғқнҷңмҠөкҙҖ көҗм •, мҠӨнғҖнӢҙ/м—җм ңнӢ°лҜёлёҢ, м§ҖлӢЁл°ұ нҳҲмһҘкөҗнҷҳмҲ (н• мҲҳ мһҲлҠ” кІҪмҡ°)мқҙ м№ҳлЈҢм—җ н•„мҲҳм ҒмқҙлӢӨ. м§ҖлӢЁл°ұ нҳҲмһҘкөҗнҷҳмҲ мқҖ 5м„ё нҳ№мқҖ лҠҰм–ҙлҸ„ 8м„ём—җ мӢңмһ‘н• кІғмқҙ к¶Ңкі лҗңлӢӨ. PCSK9 м–өм ңм ң, lomitapide, mipomersen к°ҷмқҖ мғҲлЎңмҡҙ м№ҳлЈҢм ңк°Җ 추к°Җлҗ мҲҳ мһҲлӢӨ. н•ңкөӯм—җм„ң mipomersenкіј lomitapideлҠ” мң нҶөлҗҳм§Җ м•Ҡкі мһҲмңјлӮҳ, PCSK9 м–өм ңм ң мӨ‘ evolocumabмқҖ HoFHм—җм„ң мҠ№мқёлҗҳм—ҲлӢӨ. н•ӯ ANGPTL3 н•ӯмІҙмқё evinacumabлҸ„ мқјл¶Җ көӯк°Җм—җм„ң HoFH нҷҳмһҗм—җкІҢ мҠ№мқёлҗҳм—ҲлӢӨ. лӢӨлҘё мӢ¬нҳҲкҙҖ мң„н—ҳ мҡ”мқё мЎ°м ҲлҸ„ мӨ‘мҡ”н•ҳл©°, м•„мҠӨн”јлҰ°лҸ„ кі л Өн• н•„мҡ”к°Җ мһҲлӢӨ.
нҠ№мҲҳ 집лӢЁмқҳ FH
мҶҢм•„
진лӢЁ: мҶҢм•„м—җм„ң FHлҠ” лҶ’мқҖ LDL-Cм—җ лҸҷл°ҳлҗң мЎ°кё° л°ңлі‘ кҙҖмғҒлҸҷл§Ҙ м§Ҳнҷҳ нҳ№мқҖ лҶ’мқҖ LDL-C нҳ№мқҖ мң м „ кІҖмӮ¬ м–‘м„ұмқҳ к°ҖмЎұл Ҙмқ„ кё°л°ҳмңјлЎң 진лӢЁн•ңлӢӨ[50]. мң„мҷҖ к°ҷмқҖ к°ҖмЎұл Ҙмқҙ мһҲлҠ” мҶҢм•„м—җм„ң л°ӣм•„л“Өм—¬м§ҖлҠ” кё°мӨҖ LDL-C мҲҳм№ҳлҠ” вүҘ 160 mg/dLмқҙлӢӨ. л§Ңмқј л¶ҖлӘЁ мӨ‘ н•ңмӘҪмқҙ мң м „ кІҖмӮ¬м—җ м–‘м„ұ мҶҢкІ¬мқҙ мһҲлӢӨл©ҙ, мһҗл…Җм—җм„ң 진лӢЁмқ„ мң„н•ң LDL-C мҲҳм№ҳлҠ” лҚ” лӮ®кІҢ мһЎмқ„ мҲҳлҸ„ мһҲлӢӨ. FHлЎң мқҳмӢ¬лҗҳлҠ” мҶҢм•„лҠ” 5м„ёл¶Җн„° м„ лі„ кІҖмӮ¬лҘј н•ңлӢӨ. к°ҖмЎұ лӮҙм—җ лі‘мқём„ұ лҸҢм—°ліҖмқҙк°Җ л°ңкІ¬лҗҳм—Ҳмқ„ л•Ңм—җлҠ” к°ҖлҠҘн•ҳл©ҙ мң м „ кІҖмӮ¬к°Җ к¶Ңкі лҗңлӢӨ. мӮ¬м¶ҳкё° мӢңкё°м—җ мҪңл ҲмҠӨн…ҢлЎӨ мҲҳм№ҳк°Җ ліҖн• мҲҳ мһҲкё° л•Ңл¬ём—җ FH нҷ•мқёмқ„ мң„н•ҙм„ңлҠ” мқҙ мӢңкё° мқҙнӣ„м—җ мһ¬кІҖмқ„ н•ҙм•ј н•ңлӢӨ. мҶҢм•„м—җм„ң HoFHм—җ лҢҖн•ң м„ лі„ кІҖмӮ¬лҠ” мөңлҢҖн•ң мқҙлҘё мӢңкё°м—җ н•ҙм•ј н•ңлӢӨ.
м№ҳлЈҢ: FHмқё мҶҢм•„лҠ” м Ғм Ҳн•ң мӢқмӮ¬м—җ лҢҖн•ң көҗмңЎмқҙ н•„мҡ”н•ҳл©°, 8-10м„ёл¶Җн„° мҠӨнғҖнӢҙмңјлЎң м№ҳлЈҢн•ңлӢӨ. м–ём ң м•Ҫл¬ј м№ҳлЈҢлҘј мӢңмһ‘н• м§ҖлҠ” мҶҢм•„мқҳ н‘ңнҳ„нҳ•, нҠ№нһҲ LDL-C мҲҳм№ҳлҘј кі л Өн•ҙ кІ°м •н•ҳл©°, LDL-C лӘ©н‘ңм№ҳлҠ” < 135 mg/dLмқҙлӢӨ[5]. мҠӨнғҖнӢҙ м№ҳлЈҢлҠ” м Җмҡ©лҹүмңјлЎң мӢңмһ‘н•ҳкі лӘ©н‘ңм№ҳ лӢ¬м„ұмқ„ мң„н•ҙм„ң мҰқлҹүн•ңлӢӨ. 2021л…„м—җ лҜёкөӯ мӢқн’Ҳмқҳм•Ҫн’Ҳм•Ҳм „мІӯмқҖ н•ӯ PCSK9 н•ӯмІҙмқё evolocumab [51]мқ„ 10м„ё мқҙмғҒмқё мҶҢм•„ FH нҷҳмһҗм—җм„ң лі‘н•©мҡ”лІ• м•Ҫм ңлЎң мҠ№мқён•ҳмҳҖлӢӨ.
мһ„мӢ л¶Җ
м—¬мһҗ FH нҷҳмһҗм—җм„ң н”јмһ„кіј мһ„мӢ мқҖ мЈјмҡ” мӮ¬м•Ҳмқҙл©° м Ғм Ҳн•ҳкІҢ л…јмқҳн•ҙм•ј н•ңлӢӨ. мқјл°ҳм ҒмңјлЎң нҳёлҘҙлӘ¬м ң н”јмһ„мқҖ кёҲкё°мқҙл©°, лӢӨлҘё н”јмһ„лІ•мқҙ м„ нҳёлҗңлӢӨ. мһ„мӢ мқ„ мӣҗн•ҳлҠ” м—¬мһҗ нҷҳмһҗлҠ” мғҒлӢҙмқ„ н•ҳкі мӢ¬нҳҲкҙҖ кІҖмӮ¬лҘј н•ҳлҠ” кІғмқҙ мўӢлӢӨ.
м§Җм§Ҳ мҲҳм№ҳ мЎ°м Ҳмқ„ мң„н•ҙ мғқнҷңмҠөкҙҖ көҗм •мқҙ к¶Ңкі лҗңлӢӨ. мөңк·ј мқјл¶Җ мһҗлЈҢк°Җ мҠӨнғҖнӢҙмқҳ нғңм•„ кё°нҳ• мң л°ң нҡЁкіјлҘј л¶Җм •н•ҳкі мһҲм§Җл§Ң, нғңм•„ лҮҢ м„ұмҲҷм—җ лҢҖн•ң м•Ҫк°„мқҳ нҡЁкіјлҘј л°°м ңн•ҳкё° м–ҙл өлӢӨ[52]. м“°кі мһҲлҚҳ м§Җм§Ҳк°•н•ҳ м•Ҫм ңлҠ” мһ„мӢ мқҙлӮҳ мҲҳмң лЎңл¶Җн„° 1-3лӢ¬ м „м—җ мӨ‘м§Җн• н•„мҡ”к°Җ мһҲлӢӨ[6]. мӢ¬н•ң FHмқё кІҪмҡ° лӢҙмҰҷмӮ° кІ°н•©мҲҳм§Җ к·ёлҰ¬кі /нҳ№мқҖ м§ҖлӢЁл°ұ нҳҲмһҘкөҗнҷҳмҲ мқ„ кі л Өн• мҲҳ мһҲлӢӨ[5]. к·ёл Үм§Җл§Ң м•Ҫм ң мӢңмһ‘ мӢңм җмқҙлӮҳ м№ҳлЈҢ лӘ©н‘ңм№ҳм—җ н•ҙлӢ№н•ҳлҠ” кіөмӢқм Ғ LDL-C мҲҳм№ҳлҠ” м—ҶлӢӨ.



 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






