


INTRODUCTION
Acquired amegakaryocytic thrombocytopenia (AAMT) is a rare disease characterized by severe thrombocytopenia due to a marked decrease in or absence of bone marrow megakaryocytes, in the presence of normal bone marrow cellularity, intact erythropoiesis, and granulopoiesis [1]. Various pathogenic mechanisms for AAMT have been suggested, including cellular and humoral suppression of megakaryocytic differentiation and cytogenetic abnormalities. Various treatments have been tried with varying clinical outcomes. Cyclosporine (CsA) and anti-thymocyte globulin (ATG) have been reported to be effective agents for the treatment of AAMT [2].
We report a patient with relapsed AAMT who was treated successfully with an additional course of CsA. Four years before relapse, the first remission was attained with the first course of CsA. A second course of CsA induced another remission. Our case suggests that an additional course of CsA can be considered for relapsed AAMT that previously responded to CsA.
CASE REPORT
A 36-year-old man visited our emergency department complaining of a 5-day history of oral bleeding and petechiae on the lower extremities. He had once worked as a printer dealing with organic solvents and had been treated for malaria 6 years earlier.
On physical examination, petechiae were noted on the extremities and abdominal wall and hemorrhagic bulla on the soft palate. No hepatosplenomegaly was observed. His blood pressure was 120/60 mmHg and body temperature was 36.8Ōäā. Complete blood counts revealed thrombocytopenia and a platelet count of 2 ├Ś 103/┬ĄL. Other laboratory findings are shown in Table 1. Platelet-associated and antinuclear antibodies were negative. Transfusion with platelet concentrate improved the thrombocytopenia.
Bone marrow aspiration and biopsy showed normal cellularity with few megakaryocytes. Erythropoiesis and granulopoiesis were normal (Fig. 1). A bone marrow cytogenetic study revealed a normal karyotype. AAMT was diagnosed.
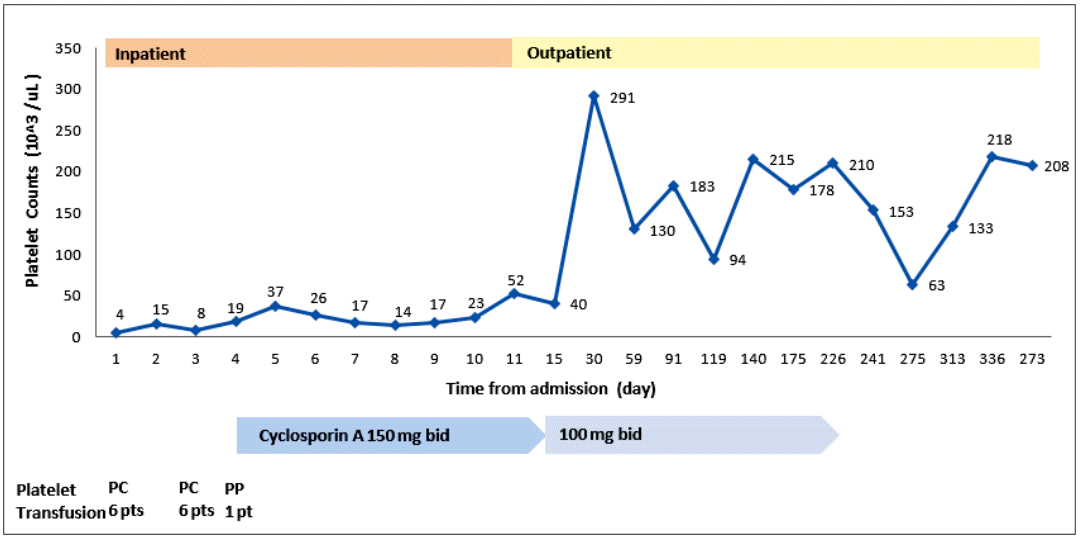
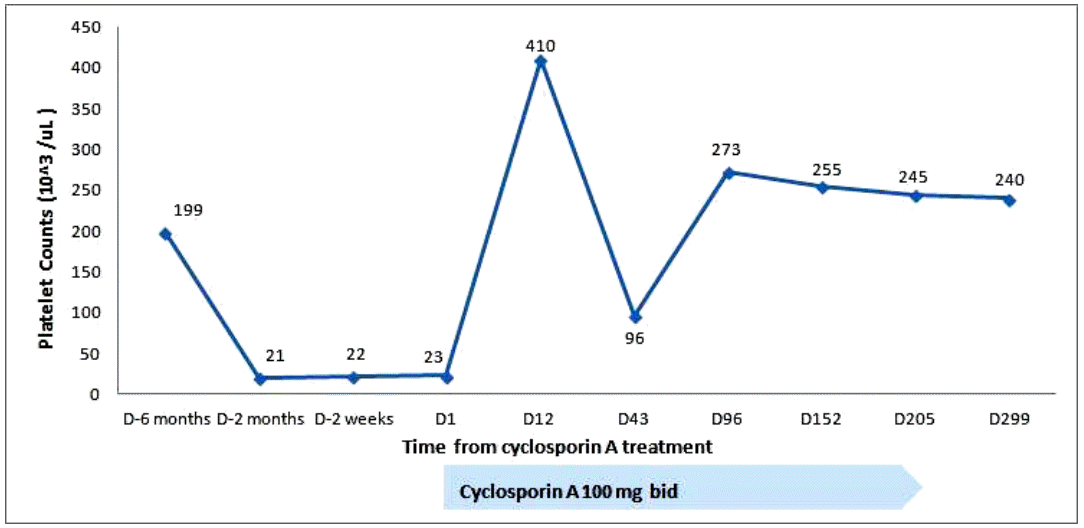
Treatment with prednisolone (1 mg/kg) was started and maintained for 2 weeks. Thereafter, it was tapered to 15 mg/day for 7 days, and then to 10 mg/day for 3 days. No improvement in the thrombocytopenia was observed. Oral bleeding with thrombocytopenia (5 ├Ś 103/┬ĄL) recurred 1 month after the diagnosis. The steroid was discontinued. Oral CsA (150 mg twice a day) was started 50 days after the diagnosis, and titrated to 100 mg twice a day to attain a serum level of CsA of 150 ng/mL. The platelet count increased progressively to 52 ├Ś 103/┬ĄL on the 11th day of CsA, and 291 ├Ś 103/┬ĄL on the 27th day. Subsequently, the serum platelet count remained at 100-200 ├Ś 103/┬ĄL (Fig. 2). We discontinued the CsA after 7 months, and the platelet count remained at 100-200 ├Ś 103/┬ĄL for the following 4 years until the thrombocytopenia (22 ├Ś 103/┬ĄL) recurred. Suspecting relapsed AAMT or progression to other hematological disorders, such as myelodysplastic syndrome (MDS) or aplastic anemia (AA), bone marrow aspiration and biopsy were performed. Rare megakaryocytes, with normal bone marrow cellularity and intact erythropoiesis, and granulopoiesis were observed. Relapsed AAMT was diagnosed. To treat the relapse, CsA (100 mg twice a day) was administered for 6 months, and the thrombocytopenia improved (240 ├Ś 103/┬ĄL). No subsequent relapse has occurred (Fig. 3).
DISCUSSION
Acquired amegakaryocytic thrombocytopenia is an unusual disease characterized by thrombocytopenia due to impaired megakaryocytopoiesis. T-cell-mediated suppression and humoral inhibition of megakaryocyte colony formation by anti-thrombopoietin (TPO) IgG antibody or anti-c-Mpl (TPO receptor) antibody have been demonstrated [1-3]. Cases with cytogenetic abnormalities, such as the Philadelphia chromosome and chromosome 5 q deletion, have been reported, which lead to MDS and leukemia. Cytokine regulation and hormonal influence on megakaryocytopoiesis have also been suggested as a possible pathogenesis [2].
Unlike immune thrombocytopenic purpura, prednisone and intravenous immunoglobulin (IVIG) are usually ineffective in AAMT, while the response to transfusion is adequate. Although no consensus has been reached on the treatment of AAMT, CsA and ATG have shown satisfactory results. Quint├Īs-Cardama [4] reported successful treatment of an AAMT case with a limited CsA course. Niparuck et al. [5] reported four cases of AAMT refractory to corticosteroid, IVIG, and cyclophosphamide. Although MDS developed in one case, three of the four cases achieved complete remission with ATG and CsA. Other therapies including danazol, cyclophosphamide, azathioprine, rituximab, and bone marrow transplantation have shown varied responses [2]. One case report described an AAMT patient who was dependent on CsA because of repeated relapse and remission [6].
Cyclosporine inhibits signal transduction pathways. It binds to intracellular receptors and immunophilins, blocking the action of calcineurin, resulting in complete block of the translocation of the cytosolic component of the nuclear factor of activated T cells (NF-AT), which eventually results in failure to activate the genes regulated by the NF-AT transcription factor, including genes encoding interleukin (IL-4), CD40 ligand, and IL-2 [7]. Therefore, CsA can improve the thrombocytopenia in AAMT by suppressing both the T-cell-mediated and humoral immunity effects on megakaryocyte colony formation.
Only a few cases of the treatment of relapsed AAMT with CsA have been reported. For AA, the British Committee for Standards in Haematology recommends a second course of ATG/CsA for a relapse after the first course of ATG/CsA [8]. A retrospective study by the United States National Institutes of Health group showed that non-responders to first-line rabbit ATG/CsA who received a second course of horse ATG/CsA had a relatively poor response rate (21% at 3 months). Therefore, the research group did not recommend a second course of ATG/CsA for patients who failed to respond to first-line rabbit ATG/CsA [9]. A prior response to ATG implies an immunological pathophysiological mechanism of marrow failure, and responsiveness to repeated treatment may be expected. We suspected a similar mechanism for the good response achieved in our patient to the second course of CsA, but this has to be studied further.
For AAMT cases refractory to CsA or ATG, and cases of relapsed or progressive AAMT in patients who are relatively young and have matched siblings, allogeneic bone marrow transplantation is considered an appropriate treatment. Alemtuzumab, a T-cell-depleting agent, improves thrombocytopenia [3]. The TPO receptor agonists eltrombopag and romiplostim also elicit satisfactory responses, but careful long-term follow-up studies to assess safety are required [10].
Our patient responded to the first 7 months of treatment with CsA, and remained in remission for 4 years. When the AAMT relapsed, a second 6-month course of CsA induced another remission. For AAMT patients who have achieved remission with CsA, CsA can be administered on the relapse of the AAMT. Regular long-term follow-up is necessary to monitor relapse or progression of AAMT.
In summary, AAMT is an unusual disease characterized by severe thrombocytopenia resulting from a marked decrease in bone marrow megakaryocytes. A few cases have been satisfactorily treated with CsA and ATG. We report a patient with relapsed AAMT who was successfully treated with a second course of CsA. For patients who have achieved remission with CsA, an additional course of CsA can be considered at the relapse of AAMT.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






