


INTRODUCTION
Systemic necrotizing vasculitis, also known as polyarteritis nodosa (PAN), primarily affects medium-sized muscular arteries. Although small muscular arteries are occasionally also affected, arterioles, venules, and capillaries are typically spared [1].
The annual incidence of PAN is < 1/million/year, but an incidence of 77/million/year has been recorded in hepatitis B virus-endemic areas, with a male to female ratio of 2-3:1 [2]. Renal involvement occurs in 80-90% of patients and is characterized by tissue infarction or hematoma, typically caused by rupture of a renal microaneurysm [3,4]. However, cases exhibiting only kidney involvement are extremely rare. Therefore, in such cases, early suspicion and diagnosis may be challenging.
Glucocorticoids and cyclophosphamide are the cornerstones of PAN therapy. The involved organs and disease progression are the two principal considerations when treating patients with PAN [5].
Given earlier diagnoses and more efficient therapies, the overall prognosis of PAN has recently improved for patients who receive treatment, and the 5-year survival rate is approximately 80% [6,7]. However, if PAN is not treated effectively, death typically results from progressive renal failure or gastrointestinal complications [8].
Here, we report the case of a 23-year-old male with isolated renal PAN who presented with proteinuria and mild renal function impairment.
CASE REPORT
In February 2019, a 19-year-old man with no specific medical or familial history was referred to our hospital for further examination following a positive proteinuria test result during an annual student medical examination. These examinations are routine in the first to third years of high school in our country.
On presentation at our hospital, he denied any systemic symptom including edema and dysuria. His physical examination findings were as follows: blood pressure 134/80 mmHg, pulse rate 84 beats/min, respiratory rate 20 breaths/min, and body temperature 36.5℃. His laboratory urinalysis results were proteinuria 30 mg/dL, no hematuria, and a urinary protein-to-urinary creatinine (Cr) ratio of 0.880 g/gCr. His blood urea nitrogen, level, blood Cr levels, and estimated glomerular filtration rate (eGFR; chronic kidney disease epidemiology collaboration [CKD-EPI] Cr equation) were 16 mg/dL, 1.13 mg/dL, and 94.4 mL/min/1.73 m2, respectively. His peripheral blood tests, hepatic function tests, complement levels, and other serological and virological tests were all normal.
No specific findings were apparent on plain chest or plain abdominal radiography. Ultrasonography indicated that the kidneys were reduced in size (right, 8.1 × 4.2 cm; left, 8.4 × 4.7 cm). On 25 February 2019, the patient underwent renal biopsy, and immunoglobulin A (IgA) nephropathy was diagnosed.
On histological examination, the patient was classified with M0, E0, S0, T0, and C0 disease according to the Oxford classification. Inflammation and necrosis involving the arterial intima and media, which are typical findings of PAN, were not histologically observed.
The patient was followed-up regularly for the proteinuria while taking losartan potassium 50 mg/T on an outpatient basis. This effectively controlled the proteinuria, and renal function was well-maintained. The patient was counselled in terms of appropriate exercise, rest, and diet during follow-up.
Subsequently, on 6 February 2023, 4 years after the diagnosis of IgA nephropathy, his Cr level was found to have increased to 1.49 mg/dL and a decreased eGFR (CKD-EPI; Cr, 59 mL/min/1.73 m2) was also observed. Given that a sudden rise in the Cr level of a patient with IgA nephropathy is atypical, the presence of co-existing kidney disease(s) was investigated. No specific findings were observed except that both kidneys remained small on renal ultrasonography, as at the time of IgA diagnosis.
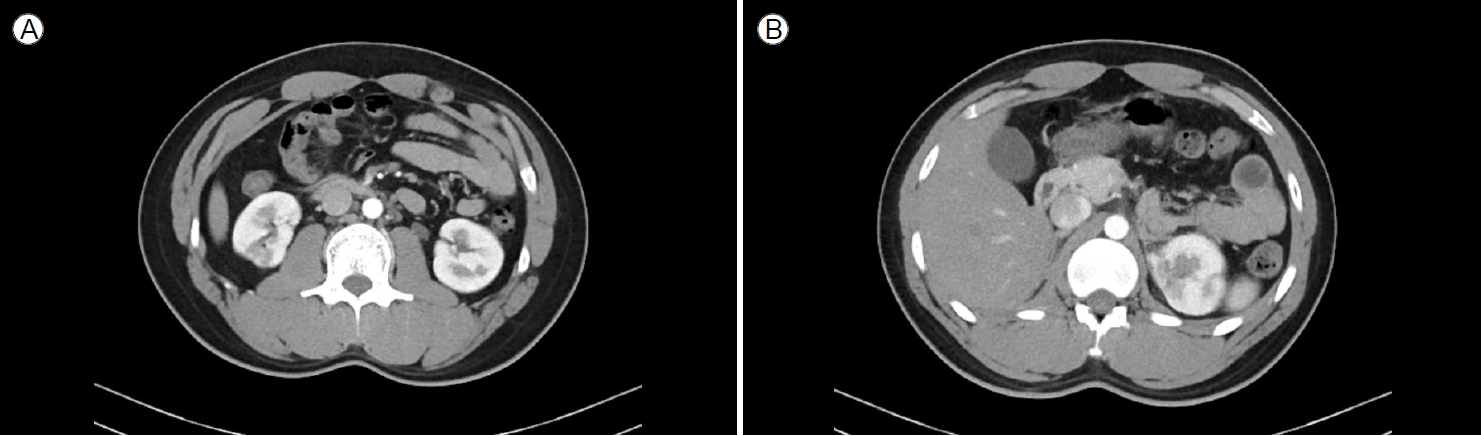
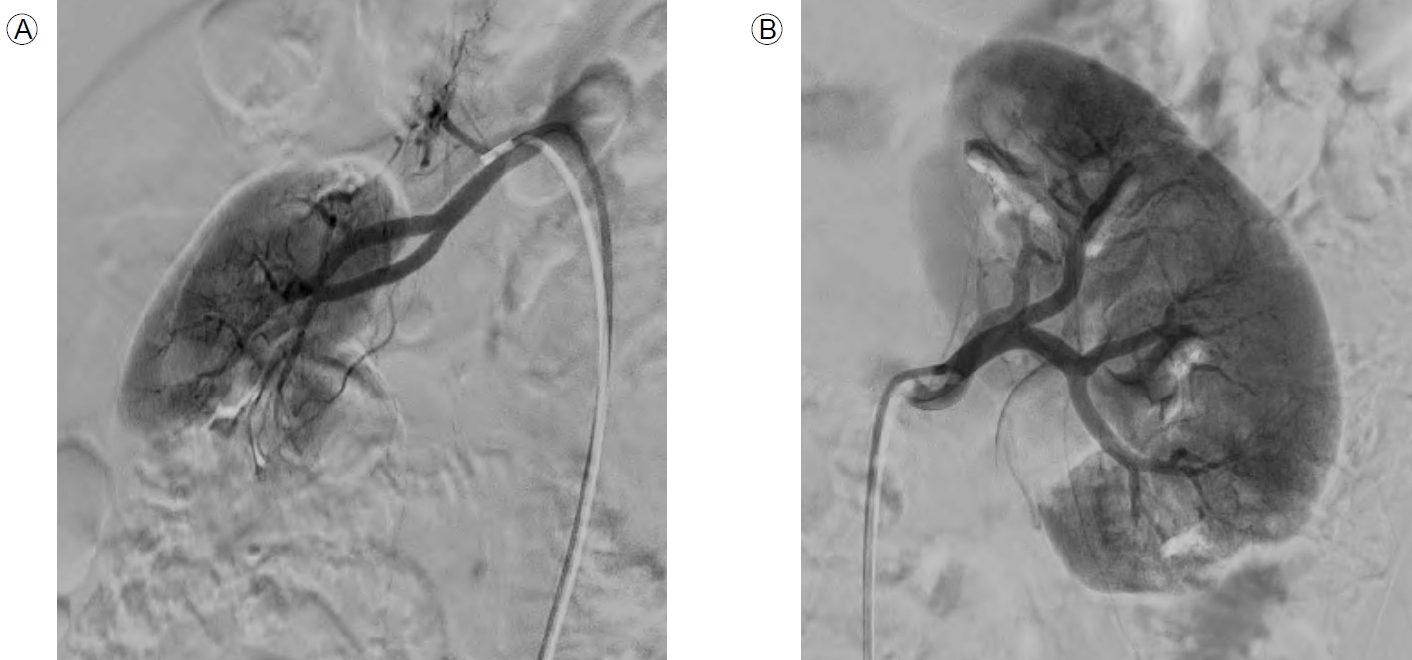
Computed tomography (CT) scans of the kidneys, ureters, and bladder (the KUB-CT set) were obtained to determine the cause of deterioration in renal function. According to the results of the KUB-CT set performed on 7 February 2023, there were cortical defects in both kidneys: a round low-attenuated lesion on the upper pole of the left kidney and a wedge-shaped low-attenuated lesion on the lower pole of the right kidney (Fig. 1). There was no clinical evidence of infection. Therefore, the multifocal cortical defects were suspected to be infarctions caused by emboli, and additional medical examinations were scheduled. A bilateral renal angiogram was performed on 21 February 2023 to explore possible abnormalities in the renal artery. The angiographic findings indicated partial obliteration or flattening of the left distal renal artery branches (from the distal renal arteries to the arcuate arteries). Multifocal extensively infarcted lesions were observed in the right kidney along with localized infarcted lesions in the upper pole of the left kidney. These findings were suggestive of occlusive disease in both kidneys, leading to a diagnosis of PAN (Fig. 2).
During an outpatient clinical visit on 28 February 2023, and based on the diagnosis of PAN, treatment was initiated with prednisolone (60 mg for 6 weeks). We performed our final examination on 7 April 2023, when the steroid treatment was tapering, and noted improvements in his Cr level (1.28 mg/dL), eGFR (CKD-EPI; Cr 78.9 mL/min/1.73 m2), and urinary protein-to-urinary Cr ratio (0.184 g/gCr).
DISCUSSION
PAN is a systemic necrotizing vasculitis that primarily affects medium-sized muscular arteries. Although small muscular arteries may be occasionally affected, arterioles, venules, and capillaries are typically spared [1]. The annual incidence is < 1/million/year, but an incidence of 77/million/year has been recorded in HBV-endemic areas, with a male/female ratio of 2-3:1 [2]. The average age at onset is approximately 50 years, and the peak incidence occurs in the fifth and sixth decades of life [9].
Here, we report a patient with vasculitis localized to the kidneys who was diagnosed with IgA nephropathy after he underwent a kidney biopsy to determine the cause of proteinuria identified during a routine high school health checkup. At that time, the pathological findings in the context of IgA nephropathy did not reveal any vascular abnormalities suggestive of PAN. Pathological findings indicative of PAN include inflammatory infiltrates that are typically mixed in nature, thus including lymphocytes, macrophages, and variable numbers of neutrophils and eosinophils [10].
Our patient was treated with angiotensin receptor blockers following the diagnosis of IgA nephropathy, after which the proteinuria improved and the renal function remained stable during the follow-up period. However, 4 years later, given a sudden and atypical rise in the Cr level, several tests were performed to investigate the possibility of co-existing kidney disease. PAN may cause a variety of clinical manifestations in a high proportion of patients, including non-specific symptoms such as weakness, fatigue, weight loss, fever, arthralgia, and myalgia, as well as symptoms indicating target organ dysfunction or damage [3,6]. However, our patient had no specific symptoms other than an elevated Cr level, and urinalysis revealed only mild proteinuria and no hematuria.
In most patients with PAN, incomplete luminal narrowing of the inflamed arteries causes glomerular ischemia but not inflammation or necrosis. Consequently, such patients frequently exhibit sub-nephrotic (i.e., minimal) proteinuria with only modest hematuria. Typically, no red blood cell casts are observed, and infarcts in the kidneys can sometimes be clinically asymptomatic [6].
PAN does not cause glomerulonephritis but frequently triggers hypertension, reflecting the effects of PAN on the intrarenal artery [3,6]. However, our patient maintained a constant blood pressure.
Any PAN diagnosis should ideally be confirmed via biopsy of the (suspected) clinically damaged organ, given the relative rarity of the disease and the possibility of serious treatment-related adverse effects. However, it has been suggested that a renal biopsy should be performed only for patients with negative arteriographic results because small microaneurysms in the kidneys may increase the risk of bleeding during biopsy. As our patient had undergone a renal biopsy at the time of diagnosis of IgA nephropathy, we considered it appropriate to obtain a KUB-CT set to detect other kidney diseases; this revealed multiple renal infarctions. Subsequently, renal angiography was performed. Partial obliteration or flattening of the left distal renal artery branches (distal to the arcuate arteries) and extensively infarcted multifocal lesions in the right and left kidneys were observed on both renal angiographic investigations. All other possible diseases were ruled out via assays of immunological markers.
The typical arteriographic lesions of PAN are arterial saccular or fusiform microaneurysms 1-5 mm in diameter, which usually co-exist with stenotic lesions. In the absence of an organ that clearly should be biopsied, angiography occasionally reveals blood vessel microaneurysms in the renal, hepatic, or mesenteric circulations [3,11].
Intimal proliferation and thickening of the inflammatory vascular wall may reduce blood flow and predispose affected vessels to thrombosis. Arterial narrowing and thrombosis aside, inflammation can weaken vessel walls; aneurysms may then form. If hypertension is present, aneurysmal rupture can cause life-threatening hemorrhage.
PAN is characterized by the presence of medium-sized vascular microaneurysms. Even in the absence of histological proof, PAN can be safely diagnosed when the distinctive angiographic changes are observed [3,12].
Our patient had radiological findings compatible with PAN. However, no aneurysm was observed; an aneurysm is a characteristic diagnostic finding of PAN. In this case, the absence of an aneurysm may reflect the fact that PAN was detected early, before an aneurysm had formed.
Glucocorticoids and cyclophosphamide are crucial in PAN treatment. The current therapeutic strategies for mild primary PAN involve the administration of corticosteroids alone [7]. The standard dosage of prednisone or prednisolone is 1 mg/kg/day, with subsequent tapering when remission is achieved [8].
In addition to corticosteroids, cyclophosphamide is administered when critical organ involvement persists [8].
Given that our patient exhibited only mild worsening of renal function and no other organ involvement, we opted to treat him with steroid monotherapy. Indeed, this successfully treated the isolated renal involvement in this patient with a mild form of PAN.
Renal failure and mesenteric, cardiac, or cerebral infarction(s) are the major complications and causes of death in patients with PAN. As healing of inflamed vessels can cause progressive narrowing of the arterial lumina and subsequent organ ischemia, such complications may develop even when the disease has not clinically manifested. The probability that tissue function can be maintained increases if treatment is started early in the course of the illness, as in this case.
PAN is usually clinically suspected based on the characteristic symptoms, physical findings, and consistent laboratory test results. However, as there are no known markers of PAN, it remains difficult to make a precise diagnosis. PAN is most commonly diagnosed in middle-aged or older adults, and the incidence rises with age, with a peak in the sixth decade of life. However, as in this case, it can also occur in children or young people, where suspicion of PAN is less likely. Therefore, to ensure diagnosis during the early stage of PAN, followed by immediate treatment with a steroid and immunosuppressive therapy, a strong index of suspicion is essential.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






