


ņä£ ļĪĀ
ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØĆ ņ×äņāü ļ│æļ”¼ĒĢÖņĀüņ£╝ļĪ£ Ļ░äņäĖĒżļé┤ ņ¦Ćņ¦łņØś ņČĢņĀüņØä ļ│┤ņØ┤ļŖö ņ¦Ćļ░®ņ”ØĻ│╝ ņØ┤ņŚÉ ņŚ╝ņ”ØņØä ļÅÖļ░śĒĢśļŖö ņåīĻ▓¼ņØä ļ│┤ņŚ¼ņżĆļŗż[1]. Ļ│╝ļÅäĒĢ£ ĒżĒÖöņ¦Ćļ░® ļ░Å ļŗ╣ņØś ņäŁņĘ©Ļ░Ć ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ņØ╝ņ░© ņ£Āļ░£ņØĖņ×ÉļĪ£ ņØ╝ļ░śņĀüņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░, ņ£Āļ│æļźĀņØ┤ ņ”ØĻ░ĆĒĢśļ®┤ņä£ ĻĄŁļé┤ņŚÉņä£ļŖö ņĢĮ 21.5% ņĀĢļÅäļź╝ ņ░©ņ¦ĆĒĢśĻ│Ā ņ׳ļŗż[2]. ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ņżæļō▒ļÅäļŖö ļŗ©ņł£ ņ¦Ćļ░®ņ”ØņŚÉņä£ ņŚ╝ņ”Ø, Ļ░äņäĖĒż ņåÉņāü ĻĘĖļ”¼Ļ│Ā ņä¼ņ£ĀĒÖöņØś ņ£Āļ¼┤ņŚÉ ļö░ļźĖ ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░äņŚ╝ņ£╝ļĪ£ ĻĄ¼ļČäĒĢĀ ņłś ņ׳Ļ│Ā, ņØ┤ ņżæ ņĢĮ 3-15%ņŚÉņä£ Ļ░äĻ▓Įļ│Ćņ”Ø ļśÉļŖö ļ¦ÉĻĖ░ Ļ░äņ¦łĒÖśņ£╝ļĪ£ ņ¦äĒ¢ēĒĢĀ ņłś ņ׳ņ£╝ļ®░, Ļ░äņĢöņØś ņŻ╝ņÜö ņøÉņØĖņØś ĒĢśļéśļĪ£ Ļ░äņŻ╝ļÉśņ¢┤ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ņ¦äĒ¢ē Ļ┤ĆļĀ© ĻĖ░ņĀäņØä ļ░ØĒ׳ļŖö Ļ▓āņØĆ ņ×äņāüņĀüņ£╝ļĪ£ ņżæņÜöĒĢ£ Ļ│╝ņĀ£ņØ┤ļŗż[3].
ņåīĒżņ▓┤(endoplasmic reticulum, ER)ļŖö ļŗ©ļ░▒ņ¦łņØś ĒĢ®ņä▒, ņĀæĒל, ļ│ĆĒśĢņØä ļŗ┤ļŗ╣ĒĢśļŖö ņżæņÜöĒĢ£ ņäĖĒżļé┤ ņåīĻĖ░Ļ┤Ćņ£╝ļĪ£, ņ╣╝ņŖśņØś ĒĢŁņāüņä▒ ņ£Āņ¦Ć ļ░Å ņŖżĒģīļĪż, ĒāäņłśĒÖöļ¼╝ ļ░Å ņ¦Ćņ¦łņØś ņāØĒĢ®ņä▒ņØä ņ£Āņ¦ĆĒĢśļŖö ļŹ░ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢśļ®░ Ļ░äņäĖĒżņŚÉ ĒÆŹļČĆĒĢśļŗż[4-6]. ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņŚÉņä£ ņ¦Ćļ░®Ļ░äņŚ╝ņ£╝ļĪ£ ņ¦äĒ¢ēļÉśļŖö ņĀĢĒÖĢĒĢ£ ĻĖ░ņĀäņØĆ ņל ļ░ØĒśĆņ¦Ćņ¦ĆļŖö ņĢŖņĢśņ¦Ćļ¦ī ņé░ĒÖöņŖżĒŖĖļĀłņŖż, ļ»ĖĒåĀņĮśļō£ļ”¼ņĢä ĻĖ░ļŖźņØ┤ņāü, ņĢäļööĒżņ╣┤ņØĖ ļ│ĆĒÖö, ņ¦Ćņ¦ł Ļ│╝ņé░ĒÖö ļō▒ņØ┤ multiple hit hypothesisļĪ£ ņŚ¼Ļ▓©ņ¦Ćļ®░, ņØ┤ļōż ņżæ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦łļōżņØ┤ ņåīĒżņ▓┤ļé┤ņŚÉ ņČĢņĀüļÉśļ®┤ņä£ ļ░£ņāØĒĢśļŖö ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż(ER stress)Ļ░Ć ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢ£ļŗżĻ│Ā ļ│┤Ļ│Ā ņ׳ļŗż[7,8]. ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ņåīĒżņ▓┤ņØś ĒĢŁņāüņä▒ņØä ļ░®ĒĢ┤ĒĢśļŖö ņāüĒā£ ņ”ē, ņ¦Ćņ¦ł ļśÉļŖö ļ│ĆĒśĢļÉ£ ļŗ©ļ░▒ņ¦ł ļō▒ņØś Ļ│╝ņ×ē ņČĢņĀü ļō▒ņŚÉ ņØśĒĢ┤ ņ£Āļ░£ļÉśĻ│Ā, ņØ┤ļĪ£ ņØĖĒĢ┤ ļ»ĖņĀæĒל ļŗ©ļ░▒ņ¦ł ļ░śņØæ(unfolded protein response, UPR)ņØ┤ ĒÖ£ņä▒ĒÖöļÉ£ļŗż. ĒÖ£ņä▒ĒÖöļÉ£ ļ»ĖņĀæĒל ļŗ©ļ░▒ņ¦ł ļ░śņØæņØĆ ņ¦äĒĢĄ ņāØļ¼╝ņŚÉņä£ ņל ļ│┤ņĪ┤ļÉ£ ĒŖ╣ļ│äĒĢ£ ņäĖĒżņŗĀĒśĖĻ▓ĮļĪ£ļĪ£, ļŗ©ļ░▒ņ¦ł ĒĢ®ņä▒ņØś ņżæļŗ©, ļ»ĖņĀæĒל ļŗ©ļ░▒ņ¦łņØś ļČäĒĢ┤, chaperoneņØś ļ░£Ēśä ļō▒ņØś ņ”ØĻ░Ćļź╝ ĒåĄĒĢśņŚ¼ ņåīĒżņ▓┤ ļé┤ņØś ņŖżĒŖĖļĀłņŖżļź╝ ņżäņØ┤Ļ│Ā ņäĖĒżņØś ņĀĢņāüņĀü ĻĖ░ļŖźņØä ĒÜīļ│ĄĒĢśļÅäļĪØ ĒĢ£ļŗż[9]. ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ļö░ļźĖ ņŗĀĒśĖņĀäļŗ¼ņ▓┤Ļ│äļŖö ņŻ╝ļĪ£ ļ╣äļ¦ī, ņ¦Ćļ░®Ļ░ä ņ¦łĒÖś ĒÖśņ×ÉņŚÉņä£ Ļ│ĄĒåĄņĀüņ£╝ļĪ£ Ļ┤Ćņ░░ļÉśļŖö ņäĖĒż ņ×Éļ®Ėņé¼(apoptosis), ņ¦Ćļ░® ļÅģņä▒(lipotoxicity), ņŚ╝ņ”Ø, ņØĖņŖÉļ”░ ņĀĆĒĢŁņä▒ņØä ņ£ĀļÅäĒĢśļŖöļŹ░, ņØ┤ ņäĖĒż ņŖżĒŖĖļĀłņŖż Ļ▓ĮļĪ£ļŖö ĻĘĖ ļ░¢ņŚÉ ņé░ĒÖöņŖżĒŖĖļĀłņŖż, ļ»ĖĒåĀņĮśļō£ļ”¼ņĢä ĻĖ░ļŖźņØ┤ņāü, Kupffer cell ļ¦żĻ░£ ņŚ╝ņ”ØņØä ņ×ÉĻĘ╣ĒĢśļŖö Ļ▓āņ£╝ļĪ£ļÅä ņĢīļĀżņĀĖ ņ׳ļŗż. ņØ┤ņŚÉ ļ│ĖĻ│ĀņŚÉņä£ļŖö ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņŚÉņä£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ņØśĒĢ£ ļ»ĖņĀæĒל ļŗ©ļ░▒ņ¦ł ļ░śņØæ Ļ┤ĆļĀ© ĻĖ░ņĀäņØä ņĢīņĢäļ│┤Ļ│Ā ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ļ░£ļ│æ ĻĖ░ņĀäņŚÉ ļīĆĒĢ┤ ĻĖ░ņłĀĒĢśĻ│Āņ×É ĒĢ£ļŗż.
ļ│Ė ļĪĀ
ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņÖĆ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæ
ņåīĒżņ▓┤ļŖö ļŗ©ļ░▒ņ¦ł ĒĢ®ņä▒ ļ░Å N-glycosylation, disulfide bond ĒśĢņä▒ņØä ĒåĄĒĢ┤ ļŗ©ļ░▒ņ¦łņØś ņłśņĀĢĻ│╝ ņĀæĒל(folding) ļō▒ņØś ņĀäņé¼ Ēøä ņłśņĀĢ(post-translational modifications)ņØä ĒĢśĻ▒░ļéś, ņ¦Ćņ¦łņØ┤ļéś ņŖżĒģīļĪżņØä ĒĢ®ņä▒ĒĢśļ®░ ņ╣╝ņŖśņĀĆņןņåīļĪ£ņä£ņØś ĻĖ░ļŖźņØä ņłśĒ¢ēĒĢ£ļŗż. ļŗ©ļ░▒ņ¦łņØś ņłśņĀĢņØ┤ļéś ņĀæĒלņØś Ļ│╝ņĀĢņØĆ ņäĖĒż ņāØņĪ┤ņŚÉ ĒĢäņłśņĀüņØ┤ļ®░, ņåīĒżņ▓┤ņŚÉņä£ļŖö ļŗ©ļ░▒ņ¦ł ĒÆłņ¦ł Ļ┤Ćļ”¼ ņĀÉĻ▓Ć(quality control checkpoint)ņØä ĒåĄĒĢ┤, ņĀüņĀłĒĢśĻ▓ī ņĀæĒ×ī ļŗ©ļ░▒ņ¦łļ¦ī ņåīĒżņ▓┤ņŚÉņä£ Ļ│©ņ¦Ćņ▓┤ļĪ£ ņÜ┤ļ░śĒĢĀ ņłś ņ׳ļÅäļĪØ ĒĢ£ļŗż[4,5,10]. ĒĢśņ¦Ćļ¦ī ņØ┤ļ¤¼ĒĢ£ Ļ│╝ņĀĢņØ┤ ņāØļ”¼ņĀü Ēś╣ņØĆ ļ│æļ”¼ņĀü ĒÖśĻ▓ĮņŚÉ ņØśĒĢ┤ ņåīĒżņ▓┤Ļ░Ć ņ▓śļ”¼ĒĢĀ ņłś ņ׳ļŖö ļŖźļĀź ņØ┤ņāüņØś ļ»Ėņä▒ņłÖ ļŗ©ļ░▒ņ¦łņØ┤ ņåīĒżņ▓┤ ļé┤ļĪ£ ņ£Āņ×ģņØ┤ ļÉśĻ▒░ļéś ņåīĒżņ▓┤ ļé┤ ņ╣╝ņŖśņØś ļČłĻĘĀĒśĢņØ┤ ņāØĻ▓╝ņØä ļĢī, ņåīĒżņ▓┤ņØś ņןņĢĀĻ░Ć ļ░£ņāØĒĢśļŖöļŹ░ ņØ┤ļź╝ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļØ╝Ļ│Ā ĒĢ£ļŗż. ņäĖĒżĻ░Ć ņŖżĒŖĖļĀłņŖż ņāüĒā£Ļ░Ć ļÉśļ®┤ ļČäļ╣äļŗ©ļ░▒ņ¦łņØ┤ļéś ņäĖĒżļé┤ļ¦ēĻ│ä ļŗ©ļ░▒ņ¦łņØś Ļ│Āņ░©ĻĄ¼ņĪ░ ĒśĢņä▒ņØ┤ ņĀĢņāüņ£╝ļĪ£ ņ¦äĒ¢ēļÉśņ¦Ć ņĢŖņ£╝ļ®░ ņäĖĒżļŖö ĻĘĖ ņ£äĻĖ░ņĀü ņāüĒÖ®ņŚÉņä£ ļ▓Śņ¢┤ļéśļĀżĻ│Ā ĒĢ£ļŗż. ņØ┤Ļ▓āņØĆ ņäĖĒżņāØļ”¼ĒĢÖņĀüņ£╝ļĪ£ ņØ╝ņ¢┤ļéśļŖö ņåīĒżņ▓┤ņØś ņĀĢņāüņĀü ĻĖ░ļŖźņØ┤ļŗż. ĻĘĖļלņä£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ļīĆĒĢ┤ UPRņØ┤ ĒÖ£ņä▒ĒÖöļÉ£ļŗż. UPRņØĆ ĒĢŁņāüņä▒ ļ░śņØæņ£╝ļĪ£ ņČöĻ░ĆņĀüņØĖ chaperonesņØä ņāØņä▒ņØä ņ£Āļ░£ĒĢśņŚ¼ ņåīĒżņ▓┤ ļé┤Ļ░Ģļé┤ņØś folding ļŖźļĀźņØä ņ”ØĻ░Ćņŗ£ĒéżĻ│Ā, ņåīĒżņ▓┤ Ļ┤ĆļĀ© ļŗ©ļ░▒ņ¦ł ļČäĒĢ┤(ER-associated protein degradation, ERAD)ļź╝ Ē¢źņāüņŗ£ĒéżĻ│Ā, ņåīĻĖ░Ļ┤ĆĻ│╝ ņäĖĒżļé┤ļĪ£ Ļ░ĆņĀĖņśżļŖö ļŗ©ļ░▒ņ¦ł ņĀäņé¼ ļ░Å ĒĢ®ņä▒ņØä ņĪ░ņ×æĒĢśņŚ¼ ņåīĒżņ▓┤ ļé┤Ļ░Ģņ£╝ļĪ£ņØś ļŗ©ļ░▒ņ¦ł ņ¦äņ×ģņØä ņżäņØĖļŗż[11].
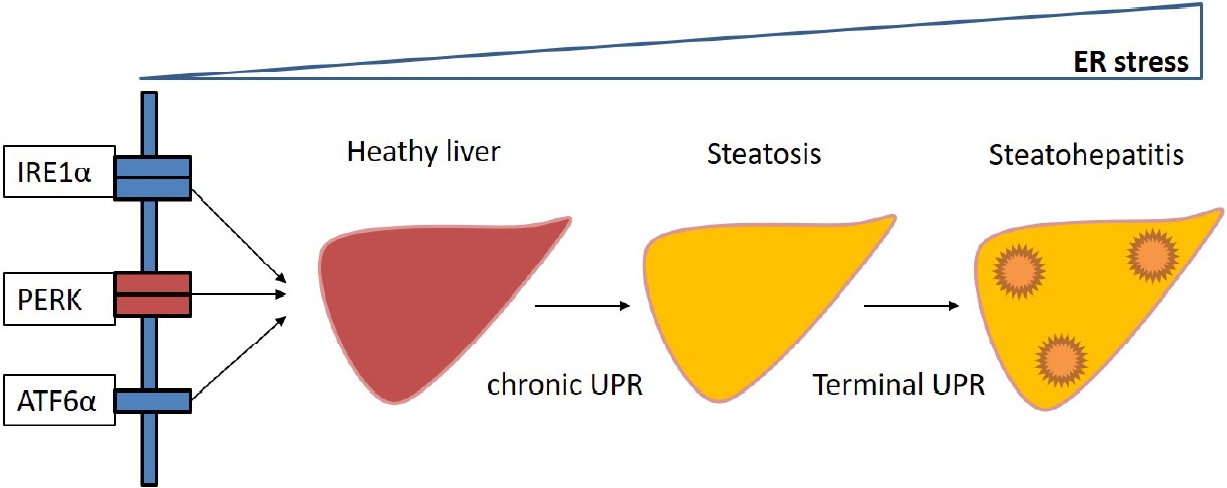
ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØś ĒÖ£ņä▒ĒÖöļŖö 3Ļ░Ćņ¦ĆņØś ņåīĒżņ▓┤ ļ¦ēĻ┤ĆĒåĄļŗ©ļ░▒ņ¦łņŚÉ ņØśĒĢ┤ ņØ┤ļŻ©ņ¢┤ņ¦äļŗż. ņåīĒżņ▓┤ ļ¦ēĻ┤ĆĒåĄļŗ©ļ░▒ņ¦łņŚÉļŖö 1) inositol-requiring enzyme 1 (IRE1), 2) PKR-like ER kinase (PERK), 3) activating transcription factor 6 (ATF6)Ļ░Ć ņ׳ļŗż(Fig. 1) [4,12]. ņĀĢņāüņĀüņØĖ ņĪ░Ļ▒┤ņŚÉņä£ļŖö ņØ┤ ļŗ©ļ░▒ņ¦łļōżņØĆ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņĪ░ņĀłņ×ÉņØĖ immunoglobulin-binding protein (BiP) ļśÉļŖö glucose-regulated protein 78 (GRP78) chaperoneĻ│╝ Ļ▓░ĒĢ®ĒĢśņŚ¼ ļ╣äĒÖ£ņä▒ĒÖö ņāüĒā£ņŚÉ ņ׳ļŗż. ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņāüĒā£ņŚÉņä£ļŖö ņØ┤ļōż chaperoneņØ┤ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņä╝ņä£ņŚÉņä£ ļČäļ”¼ļÉśļ®┤ņä£ ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻ▓ī ļÉ£ļŗż.
IRE1ņØĆ Ļ░Ćņן ņל ļ│┤ņĪ┤ļÉ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņä╝ņä£ļĪ£ isoform (IRE1╬▒, IRE1╬▓) ņżæ, IRE1╬▒ļŖö N-ļ¦Éļŗ©ņØś ņä╝ņä£ ļÅäļ®öņØĖ, ļ¦ēĻ┤ĆĒåĄ ļÅäļ®öņØĖ ĻĘĖļ”¼Ļ│Ā serine/threonine kinaseņÖĆ endoribonuclease (RNase)ļź╝ Ļ░Ćņ¦ä C-ļ¦Éļŗ© ņäĖĒżņ¦łļé┤ ļÅäļ®öņØĖņ£╝ļĪ£ ņØ┤ļŻ©ņ¢┤ņ¦ä ļ¦ēĻ┤ĆĒåĄļŗ©ļ░▒ņ¦łņØ┤ļŗż. ņåīĒżņ▓┤ļé┤ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦łņØś ņČĢņĀüņØĆ IRE1╬▒ oligomerisationĻ│╝ ņäĖĒżņ¦łļé┤ ļÅäļ®öņØĖņŚÉ ņ×ÉĻ░ĆņØĖņé░ĒÖöļź╝ ņ£ĀļÅäĒĢśņŚ¼ RNase ļÅäļ®öņØĖņØä ĒÖ£ņä▒ĒÖöņŗ£Ēé©ļŗż[13,14]. ļśÉĒĢ£ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦łņØ┤ ņ¦üņĀæņĀüņ£╝ļĪ£ IRE1╬▒ņÖĆ Ļ▓░ĒĢ®ĒĢśņŚ¼ ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻĖ░ļÅä ĒĢ£ļŗż[15]. IRE1╬▒ RNase ĒÖ£ņä▒ĒÖöļŖö XBP-1 mRNAļź╝ ļČäĒĢĀ(splicing)ņŗ£ņ╝£ ĒÖ£ņä▒ņØ┤ ņ׳ļŖö XBP-1 (spliced XBP-1, XBP1s) ļŗ©ļ░▒ņ¦łļĪ£ ļ░£ĒśäļÉĀ ņłś ņ׳ļÅäļĪØ ĒĢ£ļŗż[16,17]. XBP1sļŖö ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæ ļ░Å ERADņŚÉ ņŚ░Ļ┤ĆļÉ£ ņ£ĀņĀäņ×ÉņØś ņĀäņé¼ ņ┤ēņ¦äņ×ÉņÖĆ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņåīĒżņ▓┤ ļŗ©ļ░▒ņ¦ł foldingĻ│╝ ļČäļ╣äļź╝ Ļ░ĢĒÖöĒĢ£ļŗż[18]. Ļ▓īļŗżĻ░Ć IRE1╬▒ļŖö cell death proteaseņØś caspase family levelņØä ņĪ░ņĀłĒĢśļŖö microRNAļź╝ ļČäĒĢ┤ĒĢśņŚ¼ ĒĢŁņāüņä▒ņØä ĒÜīļ│ĄĒĢśĻ│Ā ņäĖĒż ļÅģņä▒ņØä ļ░®ņ¦ĆĒĢ£ļŗż[19]. ņäĖĒż ļ│┤ĒśĖņÖĆ ļ│äĻ░£ļĪ£ IRE1╬▒ļŖö apoptotic-signaling kinase 1 ĒÖ£ņä▒ĒÖöļź╝ ņ£ĀļÅäĒĢśņŚ¼ ņäĖĒż ņ×Éļ®Ėņé¼ņÖĆ Ļ┤ĆļĀ©ļÉ£ Jun-N terminal kinase (JNK)ņÖĆ p38 MAPKņØä ĒÖ£ņä▒ĒÖöņŗ£Ēé©ļŗż[20]. ĒÖ£ņä▒ĒÖöļÉ£ JNKļŖö ļ»ĖĒåĀņĮśļō£ļ”¼ņĢä ļ¦ēņ£╝ļĪ£ ņØ┤ļÅÖĒĢśņŚ¼ BimņØś ĒÖ£ņä▒ĒÖöņÖĆ Bcl-2ņØś ņ¢ĄņĀ£ļź╝ ņ┤ēņ¦äĒĢśņŚ¼ ņäĖĒżņé¼ļ®Ė ņ£ĀļÅäĒĢ£ļŗż[21,22]. p38 MAPKļŖö Bcl-2 ļ░£ĒśäņØä Ļ░Éņåīņŗ£Ēéżļ®┤ņä£ BimĻ│╝ DR5 ļ░£ĒśäņØä ņ”ØĻ░Ćņŗ£ņ╝£ CCAT/enhancer binding protein homologous protein (CHOP)ņØś ņĀäņé¼ņØĖņ×Éļź╝ ņØĖņé░ĒÖö ļ░Å ĒÖ£ņä▒ĒÖöĒĢ£ļŗż[23,24]. ņ¦ĆņåŹņĀüņØĖ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņāüĒÖ®ņŚÉņä£ļŖö IRE1╬▒ RNaseļŖö regulated IRE1╬▒-dependent decay (RIDD)ļź╝ ĒåĄĒĢ┤ ņåīĒżņ▓┤ņŚÉ Ļ▓░ĒĢ®ļÉ£ mRNAļōżņØä ļČäĒĢ┤ĒĢ£ļŗż. RIDDļŖö ņäĀĒāØņĀüņ£╝ļĪ£ ļŗ©ļ░▒ņ¦ł foldingņŚÉ Ļ┤ĆļĀ©ļÉ£ mRNA-encoding ļŗ©ļ░▒ņ¦łņØä Ēæ£ņĀüņ£╝ļĪ£ ĒĢ£ļŗż[1,19].
PERKņØś ĒÖ£ņä▒ĒÖöļŖö ļ▓łņŚŁĻ░£ņŗ£ņØĖņ×ÉņØĖ, eukaryotic initiation factor 2╬▒ (eIF2╬▒)ņØś ņØĖņé░ĒÖöļź╝ ņ£ĀļÅäĒĢ£ļŗż[25]. eIF2╬▒ļŖö ņåīĒżņ▓┤ņŚÉņä£ņØś ļŗ©ļ░▒ņ¦ł Ļ│╝ļČĆĒĢśļź╝ ņÖäĒÖöņŗ£ĒéżĻĖ░ ņ£äĒĢ┤ activation transcription factor 4 (ATF4)ņØś translationņØä ĒÖ£ņä▒ņŗ£ĒéżļŖö ļ░śļ®┤ņŚÉ 80s ļ”¼ļ│┤ņå£ ĒĢ®ņä▒ņØä ņ¢ĄņĀ£ĒĢ©ņ£╝ļĪ£ņŹ© mRNAņØś translationņØä ņżæļŗ©ņŗ£ņ╝£ ļŗ©ļ░▒ņ¦ł ĒĢ®ņä▒ņØä ņ¢ĄņĀ£ĒĢ£ļŗż[26,27]. ATF4ļŖö ļŗ©ļ░▒ņ¦ł folding, autophagy, redox homeostasis, ņĢäļ»ĖļģĖņé░ ļīĆņé¼ ļ░Å ņäĖĒż ņ×Éļ®Ėņé¼ņÖĆ Ļ┤ĆļĀ©ļÉ£ UPR Ēæ£ņĀü ņ£ĀņĀäņ×É ņĀäņé¼ļź╝ ĒÖ£ņä▒ĒÖöņŗ£Ēé©ļŗż[28]. ATF4ļŖö CHOP, growth arrest and DNA damage 34 (GADD34)ņÖĆ ATF3ņØä ņĪ░ņĀłĒĢśņŚ¼ ĒÖ£ņä▒ĒÖöņŗ£Ēé©ļŗż. CHOPņØĆ GADD153ņØ┤ļØ╝Ļ│Ā ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░, CCAT/enhancer binding protein (C/EBPs) ņżæ ĒĢśļéśļĪ£ CHOP ļŗ©ļ░▒ņ¦ł C-ļ¦Éļŗ© ļČĆņ£äņØś basic-leucine zipper (bZIP) ļÅäļ®öņØĖņØĆ CHOPņŚÉ ņØśĒĢ┤ ņ£ĀļÅäļÉśļŖö ņäĖĒż ņ×Éļ®Ėņé¼ņŚÉņä£ ņżæņÜöĒĢśļŗż[9]. CHOPņØś ļ░£Ēśäņ£╝ļĪ£ ņåīĒżņ▓┤ ļé┤ Ļ│ĀļČäņ×É ļŗ©ļ░▒ņ¦ł ļ│ĄĒĢ®ņ▓┤ņØś ņČĢņĀüņØä Ļ░ĆņĀĖņÖĆ ņåīĒżņ▓┤ņØś ĻĖ░ļŖźņØ┤ ņāüņŗżļÉśĻ│Ā, BAXļź╝ ņäĖĒżņ¦łņŚÉņä£ ļ»ĖĒåĀņĮśļō£ļ”¼ņĢäļĪ£ ņØ┤ļÅÖņŗ£ņ╝£ ņäĖĒż ņ×Éļ®Ėņé¼ļź╝ ņ£ĀļÅäĒĢśĻ│Ā Bcl-2ļéś BIPņØś ļ░£ĒśäņØ┤ ņ”ØĻ░ĆļÉśļŖö Ļ▓ĮņÜ░ņŚÉļŖö CHOP ļŗ©ļ░▒ņ¦ł ĒÖ£ņä▒ĒÖöĻ░Ć ņ¢ĄņĀ£ļÉ£ļŗż[29,30]. CHOPņØĆ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ņØśĒĢ┤ ļ░£ĒśäņØ┤ ņ”ØĻ░ĆļÉśļ®░, ļ¬©ļōĀ UPR ņä╝ņä£ņŚÉ ņØśĒĢ┤ ĒÖ£ņä▒ĒÖöļÉśļŖöļŹ░, ņØ┤ ņżæ PERK-eIF2╬▒-ATF4 Ļ▓ĮļĪ£Ļ░Ć CHOP ļ░£ĒśäņŚÉ ņżæņÜöĒĢśļŗż[9,30]. ņØ┤Ēøä CHOPņŚÉ ņØśĒĢ┤ ņ”ØĻ░ĆļÉ£ caspase-3, DOCs, GADD34, endoplasmic reticulum oxidoreductin 1Ļ│╝ tribbles-related protein 3ļŖö ņäĖĒż ņ×Éļ®Ėņé¼ļź╝ ņ£ĀļÅäĒĢśĻ▓ī ļÉ£ļŗż[31]. ņØ┤ļōż ņżæ GADD34ļŖö protein phosphatase 1Ļ│╝ ņāüĒśĖņ×æņÜ®ņØä ĒåĄĒĢ┤ eIF2╬▒ņØś ĒāłņØĖņé░ĒÖöļź╝ ņ£ĀļÅäĒĢśņŚ¼ ņĀäņé¼ļź╝ ņ¢ĄņĀ£ĒĢśļ®░ ĻĘĖ Ļ▓░Ļ│╝ļĪ£ ņäĖĒż ņ×Éļ®Ėņé¼ļź╝ ņ£ĀļÅäĒĢ£ļŗż[32,33].
ATF6ļŖö ņåīĒżņ▓┤ļ¦ēņŚÉ ņĪ┤ņ×¼ĒĢśļŖö ņĀäņé¼ņØĖņ×ÉļĪ£ņä£ C-ļ¦Éļŗ©ņØĆ ņåīĒżņ▓┤ ņ¬ĮņŚÉ ņ£äņ╣śĒĢśĻ│Ā, bZIPĻ│╝ DNA ņĀäņé¼ ĒÖ£ņä▒ ņśüņŚŁņØä ĒżĒĢ©ĒĢśļŖö N-ļ¦Éļŗ©ņØĆ ņäĖĒżņ¦łļé┤ņŚÉ ņ£äņ╣śĒĢ£ļŗż[9,34]. ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņāüĒÖ®ņŚÉņä£ļŖö ATF6Ļ░Ć Ļ│©ņ¦Ćņ▓┤ļĪ£ ņØ┤ļÅÖĒĢśņŚ¼ site-1 protease (S1P)ņÖĆ S2PņŚÉ ņØśĒĢ┤ ļČäĒĢ┤ņĀłļŗ©(proteolytic cleavage)ļÉ£ļŗż. ņĀłļŗ©ļÉ£ Nļ¦Éļŗ© ATF6ļŖö ĒĢĄņ£╝ļĪ£ ņØ┤ļÅÖĒĢśņŚ¼ ATF/cAMP-response elementsņÖĆ ER stress response elementsņÖĆ Ļ▓░ĒĢ®ĒĢśņŚ¼ ļŗ©ļ░▒ņ¦ł foldingņŚÉ ņżæņÜöĒĢ£ ĒÜ©ņåīņÖĆ chaperoneņØś ļ░£ĒśäņØä ņ£ĀļÅäĒĢ£ļŗż[11,35,36].
ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņÖĆ ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖś
ņåīĒżņ▓┤ļŖö ļŗ©ļ░▒ņ¦ł ņ▓śļ”¼ ņÖĖņŚÉļÅä Ļ░äņäĖĒżļé┤ņØś ņ¦Ćļ░® ĒĢ®ņä▒ņØś ņżæņÜö ĻĖ░Ļ┤Ćņ£╝ļĪ£ ņŻ╝ņÜö ņ¦Ćņ¦ł ļīĆņé¼ Ļ▓ĮļĪ£(ņ¦Ćļ░®ņāØņä▒, ņżæņä▒ņ¦Ćļ░® ĒĢ®ņä▒, ļ░Å ņĀĆņן, ņĢäĒżņ¦Ćņ¦łļŗ©ļ░▒ņ¦łņØś ĒĢ®ņä▒ ļ░Å ļČäļ╣ä, ņ¦Ćļ░®ņé░ ņé░ĒÖö)ņŚÉ Ļ┤ĆļĀ©ļÉśņ¢┤ ņ׳ļŗż. ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņŚÉņä£ļŖö ņ¦Ćļ░®ņé░Ļ│╝ ņĮ£ļĀłņŖżĒģīļĪż ļō▒ņŚÉ ņ£ĀļÅäļÉ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżĻ░Ć ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØä ĒåĄĒĢ┤ Ļ░äļé┤ ņ¦Ćņ¦ł ļīĆņé¼ ļ░Å ņ¦Ćļ░®ļÅģņä▒Ļ│╝ ņŚ░Ļ┤ĆļÉśņ¢┤ ņ¦łļ│æņØś ņ¦äĒ¢ēņØä ņ£Āļ░£ĒĢ£ļŗżĻ│Ā ļ│┤Ļ│Āņ׳ļŗż[37].
ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ļ░£ņāØņØś ņ▓½ ļŗ©Ļ│äļŖö Ļ░äņäĖĒżļé┤ ņ¦Ćņ¦ł ļ░®ņÜĖņØś ņČĢņĀüņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢśļŖö Ļ░äņ¦Ćļ░®ņ”ØņØ┤ļŗż. ņØ┤ņÖĆ ņŚ░Ļ┤ĆļÉ£ ņåīĒżņ▓┤ņØś ņŚŁĒĢĀņØä ņé┤ĒÄ┤ļ│┤ļ®┤, ņ▓½ņ¦Ė, de novo lipogenesisĻ░Ć ņåīĒżņ▓┤ ļ¦ēņŚÉ ņ£äņ╣śĒĢ£ ņĀäņé¼ņØĖņ×ÉļōżņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉ£ļŗż. ņØ┤ ņżæ sterol regulatory element-binding proteins (SREBPs)ļŖö ņ¦Ćļ░® ĒĢŁņāüņä▒(lipid homeostasis)ņØś ņżæņÜö ņĪ░ņĀłņØĖņ×Éļ®┤ņä£, de novo lipogenesisņŚÉ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢ£ļŗż[38]. SREBPsļŖö basic-helix loop-helix-leucine zipper ņĀäņé¼ņØĖņ×ÉļĪ£ ļ╣äĒÖ£ņä▒ĒÖö ņĀäĻĄ¼ņ▓┤ļĪ£ ņåīĒżņ▓┤ļ¦ēņŚÉ Ļ▓░ĒĢ®ļÉśņ¢┤ ņ׳ļŗż. SREBP-1ņØĆ ņ¦Ćļ░®ņé░Ļ│╝ ņżæņä▒ņ¦Ćļ░® ļīĆņé¼ļź╝ ņĪ░ņĀłĒĢśļ®░, SREBP-2ļŖö cholesterol ļīĆņé¼ ļ░Å LDL ņłśņÜ®ņ▓┤ ļ░£ĒśäņØä ņĪ░ņĀłĒĢ£ļŗż[39,40]. SREBP ĒÖ£ņä▒ĒÖöļŖö ņåīĒżņ▓┤ļé┤ņŚÉņä£ SREBP cleavage-activating protein (SCAP)Ļ│╝ insulin regulated proteinņØĖ INSIGsĻ░äņØś ļ░śņØæņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉ£ļŗż[41]. Sterol ņłśņ╣śĻ░Ć ļé«ņĢäņ¦Ćļ®┤, INSIGsĻ░Ć SCAPņŚÉņä£ ļČäļ”¼ļÉśĻ│Ā, SCAP-SREBPĻ░Ć Ļ│©ņ¦Ćņ▓┤ļĪ£ ņØ┤ļÅÖĒĢśĻ▓ī ļÉśņ¢┤ NH2-terminal regionņØä ļČäļ”¼ĒĢśļŖö S1PņÖĆ S2PņŚÉ ņØśĒĢ┤ SREBPĻ░Ć ĒÖ£ņä▒ĒÖöļÉ£ļŗż[42,43]. ĒÖ£ņä▒ĒÖöļÉ£ SREBPļŖö ĒĢĄņ£╝ļĪ£ ņØ┤ļÅÖĒĢśņŚ¼ ņŖżĒģīļĪż ņĪ░ņĀłņÜöņåī(sterol regulatory element)ņÖĆ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņ¦Ćļ░® ļīĆņé¼ļź╝ ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ ņ¦Ćļ░®ņØś ņČĢņĀüņØä ņ£ĀļÅäĒĢ£ļŗż[44,45]. ļæśņ¦ĖļĪ£ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ĒŖ╣ņ¦ĢņØĖ Ļ░äņäĖĒżļé┤ ņżæņä▒ņ¦Ćļ░®ņØä ĒżĒĢ©ĒĢ£ ņ¦Ćļ░®ļ░®ņÜĖ ĒśĢņä▒(lipid droplet formation)ņØ┤ļŗż[46]. Ļ░äņäĖĒżļŖö ņåīĒżņ▓┤ņŚÉ ņ£äņ╣śĒĢ£ acyltransferase enzyme (diacylglycerol acyltransferase, DGAT)ņŚÉ ņØśĒĢ┤ ņ¦Ćļ░®ņé░Ļ│╝ glycerolņŚÉ ņØśĒĢ┤ ĒĢ®ņä▒ļÉ£ ņżæņä▒ņ¦Ćļ░® ĒśĢĒā£ļĪ£ ņ¦Ćņ¦łņØä ņĀĆņןĒĢ£ļŗż[47]. ņżæņä▒ņ¦Ćļ░®Ļ│╝ cholesterol esterļĪ£ ĻĄ¼ņä▒ļÉ£ ņ¦Ćļ░®ļ░®ņÜĖņØĆ ņ£Āļ”¼ ņ¦Ćļ░®ņé░Ļ│╝ cholesterolņØś ņČĢņĀüņ£╝ļĪ£ ņØĖĒĢ£ ļÅģņä▒ņØä ļ¦ēĻĖ░ ņ£äĒĢ£ ņĀĆņןĻ│Ā ņŚŁĒĢĀņØä ĒĢ£ļŗż[48]. ļ¦ēņØĖņ¦Ćņ¦łļÅä ļśÉĒĢ£ ņåīĒżņ▓┤ņŚÉ ņ£äņ╣śĒĢ£ ĒÜ©ņåīļōżņŚÉ ņØśĒĢ┤ ĒĢ®ņä▒ļÉ£ļŗż[49]. ļ¦łņ¦Ćļ¦ēņ£╝ļĪ£ Ļ░äņäĖĒżļŖö ņåīĒżņ▓┤ņŚÉņä£ ĒĢ®ņä▒ļÉ£ very low density lipoprotein (VLDL)ņØä Ļ│©ņ¦Ćņ▓┤ļź╝ ĒåĄĒĢ┤ ļ░░ņČ£ĒĢ£ļŗż. ņåīĒżņ▓┤ ļé┤Ļ░Ģļé┤ņŚÉņä£ ņżæņä▒ņ¦Ćļ░®ņØ┤ VLDLļĪ£ packaging Ļ│╝ņĀĢņØ┤ ņ¦äĒ¢ēļÉśļŖöļŹ░, ņØ┤ Ļ│╝ņĀĢņŚÉ Apolipoprotein B100 (ApoB100)Ļ░ÖņØĆ ņĢäĒżņ¦Ćņ¦łļŗ©ļ░▒ņ¦łņØś ĒĢ®ņä▒ņØ┤ ĒĢäņÜöĒĢśļ®░, ņåīĒżņ▓┤Ļ░Ć ņĢäĒżņ¦Ćņ¦łļŗ©ļ░▒ņ¦łņØś ļ▓łņŚŁņØä ņżäņØ┤Ļ▒░ļéś ļČäļ╣äļź╝ Ļ░Éņåīņŗ£ņ╝£ ņ¦Ćļ░®ņ”ØņØä ņ£ĀļÅäĒĢ£ļŗż. ļśÉĒĢ£, ņåīĒżņ▓┤ chaperone ļŗ©ļ░▒ņ¦łņØ┤ ApoBņÖĆ ņŚ░Ļ┤ĆļÉśņ¢┤ ApoB ņĀæĒל, ņ¦Ćņ¦łĒÖö ļśÉļŖö Apo B co-translation ļśÉļŖö post-translational degradationņØä ļ¦żĻ░£ĒĢ£ļŗż[50]. ĻĘĖļלņä£ Ļ░äļé┤ VLDLņØ┤ Ļ│╝ņāØņä▒ļÉśļ®┤ ņØ┤ņāüņ¦Ćņ¦łĒśłņ”ØņØ┤ ņ£Āļ░£ļÉ£ļŗż. ņØ┤ļĀćļō» Ļ░äņäĖĒżļé┤ ņåīĒżņ▓┤ ĒĢŁņāüņä▒ņØĆ ņäĖĒżļ¦ē ņ¦Ćņ¦łņĪ░ņä▒ ņ£Āņ¦ĆņÖĆ Ļ░äļé┤ ļ░Å Ēśłņżæ ņ¦Ćņ¦ł ĒĢŁņāüņä▒ņØä ņ£Āņ¦Ćļź╝ ņĪ░ņĀłĒĢśļŖö ļŹ░ ņżæņÜöĒĢśļŗż. ļ¦īņä▒ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ņ¦üņĀæņĀüņ£╝ļĪ£ļŖö de novo lipogenesisņØä ņ£ĀļÅäĒĢśĻ│Ā, Ļ░äņĀæņĀüņ£╝ļĪ£ VLDL ļ░░ņČ£ņØś ļ│ĆĒÖö, ĻĘĖ ļ░¢ņŚÉ ņØĖņŖÉļ”░ ņŗĀĒśĖ ļ░Å autophagyļź╝ ĒåĄĒĢ┤ Ļ░äļé┤ ņ¦Ćņ¦ł ļīĆņé¼ņŚÉ ņśüĒ¢źņØä ņżĆļŗż. ļśÉĒĢ£ ņØ┤ņÖĆ ļ░śļīĆļĪ£ ņ”ØĻ░ĆļÉ£ Ļ░äļé┤ ņ¦Ćņ¦ł ņČĢņĀüņØ┤ ļ¦īņä▒ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļź╝ ņ£ĀļÅäĒĢśĻĖ░ļÅä ĒĢ£ļŗż. ĻĘĖļ¤¼ļ»ĆļĪ£ ņåīĒżņ▓┤ņØś ņøÉĒÖ£ĒĢ£ ĻĖ░ļŖźņØ┤ Ļ░äņäĖĒżņØś ņ¦Ćņ¦ł ļīĆņé¼ņŚÉ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢśĻ│Ā, ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ņØśĒĢ£ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØ┤ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ļ░£ņāØ ĻĖ░ņĀä ņżæ ĒĢśļéśļØ╝Ļ│Ā ņāØĻ░üĒĢĀ ņłś ņ׳ļŗż.
PERK-ATF3 pathway
PERK-eIF2╬▒-ATF4 Ļ▓ĮļĪ£ļŖö ņ¦Ćļ░® ņāØņä▒Ļ│╝ Ļ░äļé┤ ņ¦Ćļ░®ņ”ØņØä ņĪ░ņĀłĒĢ£ļŗż. PERK ņ¢ĄņĀ£ļŖö ņ¦Ćļ░®ņāØņä▒ĒÜ©ņåīņØĖ fatty acid synthase (FAS), ATP-citrate lyase, stearoyl CoA desaturase 1 (SCD1)ņØś ļ░£ĒśäņØä ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņ¢ĄņĀ£ĒĢ£ļŗż[51]. ņĄ£ĻĘ╝ ņŚ░ĻĄ¼ņŚÉ ļö░ļź┤ļ®┤ ĒĢŁņĀĢņŗĀļ│æņĢĮņĀ£ņŚÉ ņØśĒĢ£ PERK-eIF2╬▒ ņŗĀĒśĖņ▓┤Ļ│äņØś ĒÖ£ņä▒ĒÖöļŖö SREBP-1cņÖĆ SREBP-2ņØś Ļ░äņäĖĒżļé┤ ĒÖ£ņä▒ĒÖöļź╝ ĒåĄĒĢ┤ ņäĖĒżļé┤ ņ¦Ćņ¦ł ņČĢņĀüņØä ņ”ØĻ░Ćņŗ£Ēé©ļŗż[52]. GADD34ņØś ļ░£ĒśäĻ░ĢĒÖöļŖö ļīĆņé¼Ļ│╝ņĀĢņØä ļ│ĆĒÖöņŗ£ņ╝£ Ļ│Āņ¦Ćļ░®ņŗØņØ┤ņŚÉ ņ£ĀļÅäļÉśļŖö Ļ░äļé┤ ņ¦Ćļ░®ņ”ØņØä Ļ░Éņåīņŗ£Ēé©ļŗż[53].
eIF2╬▒ ņØĖņé░ĒÖöņØś Ļ░ÉņåīļŖö ņ¦Ćļ░®ĒśĢņä▒ ĒĢĄņłśņÜ®ņ▓┤ņØĖ peroxisome proliferator-activated receptor (PPAR╬│)ņÖĆ ņĀäņé¼ņØĖņ×ÉņØĖ C/EBP╬▒ņÖĆ C/EBP╬▓ (PPAR╬│ņØś ņāüņ£ä ņĪ░ņĀłņ×É)ņØś ļ░£ĒśäņØä Ļ░Éņåīņŗ£ĒéżļŖö ļŹ░ ņŚ░Ļ┤ĆļÉśņ¢┤ ņ׳ļŗż[53]. ATF4ļŖö eIF2╬▒ ņØĖņé░ĒÖöņŚÉ ņØśĒĢ┤ translation ļÉśļŖöļŹ░, Ļ│Āņ¦Ćļ░®ņŗØņØ┤ņŚÉ ņØśĒĢ£ ATF4 knockout ļ¦łņÜ░ņŖż ļ¬©ļŹĖņŚÉņä£ Ļ░äņŚÉņä£ ņ£ĀņØśĒĢ£ ņ¦Ćļ░®ĒśĢņä▒ ņ£ĀņĀäņ×Éļōż(PPAR╬│, SREBP1c, acetyl CoA carboxylase [ACC]), FASņØś ļ░£ĒśäņØä Ļ░Éņåīņŗ£ĒéżĻ│Ā, Ļ│Āņżæņä▒ ņ¦Ćņ¦łĒśłņ”Ø ļ░Å ņ¦Ćļ░®ņ”ØņØ┤ ņ¢ĄņĀ£ļÉśļŖö Ļ▓āņØä ļ│┤ņŚ¼ņżĆļŗż[54,55]. ņåīĒżņ▓┤ ļé┤Ļ░Ģļé┤ņŚÉņä£ VLDLņ£╝ļĪ£ packaging Ļ│╝ņĀĢņØ┤ ņ¦äĒ¢ēļÉśļŖöļŹ░, ņØ┤ Ļ│╝ņĀĢņŚÉ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ņØ┤ PERK-ATF4ņŚÉ ņØśĒĢ┤ ApoB100ņØś ļ▓łņŚŁņØä ņżäņØ┤Ļ▒░ļéś ļČäļ╣äļź╝ Ļ░Éņåīņŗ£ņ╝£ ņ¦Ćļ░®ņ”ØņØä ņ£ĀļÅäĒĢ£ļŗż[56]. Ļ▓īļŗżĻ░Ć ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö VLDL ņłśņÜ®ņ▓┤ņØś ļ░£Ēśä ņ”ØĻ░Ćļź╝ ĒåĄĒĢ┤ Ļ░äņ¦Ćļ░®ņ”ØņØä ņ£ĀļÅäĒĢ£ļŗż[57].
IRE1a-XBP1 pathway
ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņĪ░Ļ▒┤ņŚÉņä£ IRE1╬▒-XBP1 Ļ▓ĮļĪ£ ļśÉĒĢ£ Ļ░äļé┤ ņ¦Ćņ¦łļīĆņé¼ļź╝ ņ£Āņ¦ĆĒĢśļŖö ļŹ░ ņżæņÜöĒĢ£ ņÜöņåīļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. IRE1╬▒ļŖö ApoBļź╝ ĒżĒĢ©ĒĢ£ ņ¦Ćņ¦łļŗ©ļ░▒ņ¦łņØä ĒĢ®ņä▒ĒĢśļŖö ļŹ░ ĒĢäņÜöĒĢśļŗż. IRE1╬▒ļŖö ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ņāüĒÖ®ņŚÉņä£ Ļ░äļé┤ ņ¦Ćņ¦ł ņČĢņĀüņØä ņ¢ĄņĀ£ĒĢ£ļŗżļŖö ņé¼ņŗżņØä ĻĘ╝Ļ▒░ļĪ£ IRE1╬▒ ņ¢ĄņĀ£ ņŗ£, Ļ░äļé┤ ņ¦Ćļ░®ņ”Ø ņ”ØĻ░Ć ļ░Å Ēśłņżæ ņ¦Ćņ¦łņØ┤ Ļ░ÉņåīļÉśļŖö ņåīĻ▓¼ņØä ļ│┤ņØĖļŗż[58]. XBP1ļŖö ņ¦Ćļ░®ĒśĢņä▒ ņ£ĀņĀäņ×Éļōż(SCD1, DGAT2, ACC2)ņØś promoterņŚÉ ņ¦üņĀæ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņĀäņé¼ļź╝ ĒÖ£ņä▒ņŗ£Ēé┤ņ£╝ļĪ£ņŹ© Ļ░äļé┤ ņ¦Ćļ░®ņāØņä▒ņØä ņ£ĀļÅäĒĢ£ļŗż[59]. ĻĘĖļלņä£ XBP1 ņ¢ĄņĀ£ļŖö Ļ░äļé┤ ņ¦Ćļ░® ņŗĀņāØņä▒ņØä Ļ░Éņåīņŗ£Ēé┤ņ£╝ļĪ£ņŹ© ņ£ĀņØśĒĢśĻ▓ī ņżæņä▒ņ¦Ćļ░®, ņĮ£ļĀłņŖżĒģīļĪż, ņ¦Ćļ░®ņé░ ņłśņ╣śĻ░Ć Ļ░ÉņåīĒĢ©ņØä ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŗż. ļśÉĒĢ£ IRE1╬▒-XBP1 Ļ▓ĮļĪ£ļŖö protein disulfide isomerase ļ░£Ēśä ņ£ĀļÅäļź╝ ĒåĄĒĢ┤ Ļ░äļé┤ VLDL ĒĢ®ņä▒Ļ│╝ ļ░░ņČ£ņØä ņĪ░ņĀłĒĢśļŖö ļŹ░ ĒĢäņłśņĀüņØĖ ņŚŁĒĢĀņØä ĒĢśļŖö microsomal TG transport protein ĒÖ£ņä▒ļÅäļź╝ ņ”ØĻ░Ćņŗ£Ēé©ļŗż[60].
ATF6 pathway
ATF6 ņŚŁņŗ£ ņ¦Ćņ¦ł ņČĢņĀüņŚÉ ņŚŁĒĢĀņØä ĒĢśļŖöļŹ░, SREBPsņÖĆ Ļ░ÖņØĆ Ļ│©ņ¦Ćņ▓┤ ļé┤ S1PņÖĆ S2PņŚÉ ņØśĒĢ┤ ĒÖ£ņä▒ĒÖöļÉ£ļŗż[36]. ATF6╬▒ļŖö PPAR╬▒ņÖĆņØś ņāüĒśĖ ņ×æņÜ®ņØä ĒåĄĒĢ┤ Ļ░äļé┤ ņ¦Ćļ░®ņé░ņØś ņé░ĒÖöļź╝ ņ£ĀļÅäĒĢ£ļŗż. ņŗØņØ┤ņ£ĀļÅä ņØĖņŖÉļ”░ ņĀĆĒĢŁņä▒ ņźÉļ¬©ļŹĖņŚÉņä£ ATF6╬▒ļŖö PPAR╬▒/retinoid X receptor complexļź╝ ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ Ļ░äņäĖĒżļé┤ ņ¦Ćļ░®ņé░ ņé░ĒÖöņŚÉ Ļ┤ĆĻ│äļÉ£ ņ£ĀņĀäņ×É(carnitine palmitoyltransferase 1A, medium-chain acyl-coenzyme A dehydrogenase, fibroblast growth factor 21) ņĀäņé¼ ĒÖ£ņä▒ĒÖöļź╝ ņŗ£ņ╝£ Ļ░äļé┤ ņ¦Ćļ░®ņ”ØņØś ļ░£ņāØņØä ņ¢ĄņĀ£ĒĢ£ļŗż[61]. ATF6 Ļ│╝ļ░£ĒśäņØĆ SREBP2ņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉśļŖö ņ¦Ćļ░®ĒśĢņä▒ ņ£ĀņĀäņ×ÉļōżņØś ņĀäņé¼ ļ░Å ņ¦Ćņ¦ł ņČĢņĀüņØä ņ¢ĄņĀ£ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀĒĢ£ļŗż. ATF6╬▒ knockout ļ¦łņÜ░ņŖżņŚÉņä£ļŖö Fas ╬▓-oxidation ļ░Å VLDL ĒśĢņä▒ņØä ņ¢ĄņĀ£ĒĢśņŚ¼ Ļ░äļé┤ ņ¦Ćļ░®ņØ┤ ņ”ØĻ░ĆĒĢśĻ▓ī ĒĢ£ļŗż[62]. Ļ│Āņ¦Ćļ░®ņŗØņØ┤ ņ£ĀļÅä AFT6╬▒-/- ļ¦łņÜ░ņŖż ņŚ░ĻĄ¼ņŚÉņä£ļÅä SREBP-1c ļ░£Ēśä ņ”ØĻ░ĆņÖĆ ĒĢ©Ļ╗ś Ļ░äļé┤ ņ¦Ćļ░®ņ”Ø ļ░Å ļŗ╣ļČłļé┤ņä▒ņØ┤ ļ░£ņāØĒĢśļŖö Ļ▓āņØä ļ│┤Ļ│ĀĒĢ£ļŗż[63].
ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░äņŚ╝ņŚÉņä£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ņØśĒĢ£ ņŚ╝ņ”Ø ļ░śņØæ
ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØĆ ļŗżņ¢æĒĢ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ļīĆĒĢ┤ņä£ Ļ░äņäĖĒżļź╝ ļ│┤ĒśĖĒĢ©ņ£╝ļĪ£ņŹ© Ļ░äĻĖ░ļŖźņØś ĒĢŁņāüņä▒ņØä ņ£Āņ¦ĆĒĢśļŖö Ļ▓āņØä ļ¬®ņĀüņ£╝ļĪ£ ĒĢ£ļŗż. ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņŚÉņä£ļŖö ĻĖēņä▒ ļśÉļŖö ļ¦īņä▒ņĀüņØĖ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņŚÉ ļīĆĒĢ┤ ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØ┤ ņŖżĒŖĖļĀłņŖżļź╝ ņÖäĒÖöņŗ£Ēéżņ¦Ć ļ¬╗ĒĢśĻ▓ī ļÉśļ®┤, ņåīĒżņ▓┤ ĒĢŁņāüņä▒ņØä ņ×āĻ▓ī ļÉśņ¢┤ terminal UPRļĪ£ ļ│ĆĒÖśļÉśņ¢┤ reactive oxygen species (ROS) ņāØņé░, BCL-2 family ņāüĒ¢źņĪ░ņĀł, microRNA ņĪ░ņĀł, ņ¦ĆņåŹņĀüņØĖ ņåīĒżņ▓┤ ņ╣╝ņŖś ļ░░ņČ£ ļō▒ņØś ļŗżņ¢æĒĢ£ Ļ▓ĮļĪ£ļōżņØä ĒåĄĒĢ┤ ņŚ╝ņ”Ø ļ░Å ņäĖĒż ņé¼ļ®ĖņØä ņ£ĀļÅäĒĢśĻ▓ī ļÉśņ¢┤ ļŹö ņżæņ”Ø ņ¦łĒÖśņ£╝ļĪ£ ņ¦äĒ¢ēĒĢśĻ▓ī ļÉ£ļŗż(Fig. 2) [64,65].
ņØ┤ņÖĆ Ļ┤ĆļĀ©ļÉśņ¢┤ Ļ░äņäĖĒż ļé┤ņŚÉņä£ ņ”ØĻ░ĆļÉ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæ ņŗĀĒśĖņ▓┤Ļ│äļź╝ ĒåĄĒĢ┤ CHOPņØä ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ ņäĖĒż ņ×Éļ®Ėņé¼ļź╝ ņ£ĀļÅäĒĢśĻ│Ā ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ņ¦äĒ¢ēņØä ņ£ĀļÅäĒĢ£ļŗż. ņ¦Ćļ░®Ļ░äņŚ╝ ĒÖśņ×ÉņØś Ļ░äņĪ░ņ¦ü Ļ▓Ćņé¼ņāü ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ļ¦łņ╗żņØĖ CHOP, GRP78ņØś ņāüņŖ╣ ņåīĻ▓¼Ļ│╝ CHOP/GRP78 ļ╣äņ£©ņØś ļ│ĆĒÖöĻ░Ć ņ¦Ćļ░®Ļ░ä ņ¦łĒÖś ĒÖ£ņä▒ļÅä ņĀÉņłśņÖĆ Ļ░äņåÉņāüĻ│╝ ņ¢æņØś ņāüĻ┤ĆĻ┤ĆĻ│äĻ░Ć ņØ┤ļź╝ ņל ļ│┤ņŚ¼ņżĆļŗż[66,67]. ĒżĒÖö ņ¦Ćļ░®ņé░ ņżæņØś ĒĢśļéśņØĖ palmitic acidņŚÉ ņ£Āļ░£ļÉ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżĻ░Ć IRE1╬▒ Ļ▓ĮļĪ£ļź╝ ĒåĄĒĢ┤ JNK/CHOP activityļź╝ ņ£ĀļÅäĒĢśņŚ¼ BH3-only proteins PUMA, BIM ļ░£ĒśäņŚÉ Ļ┤ĆņŚ¼ĒĢśļ®░ BAXļź╝ ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻ│Ā, Ļ░äņäĖĒż ņé¼ļ®ĖņØä ņ£ĀļÅäĒĢ£ļŗż[68]. Ļ│Āņ¦Ćļ░®ņŗØņØ┤ļĪ£ ņ£ĀļÅäļÉ£ ļ¦łņÜ░ņŖż ļ¬©ļŹĖņŚÉņä£ļÅä CHOP ļ░£Ēśä ļ░Å ņäĖĒż ņ×Éļ®Ėņé¼ņÖĆ Ļ┤ĆļĀ©ļÉ£ caspase-3Ļ░Ć ĒÖ£ņä▒ĒÖöļÉ©ņØä ĒåĄĒĢ┤ ņ£ä ņé¼ņŗżņØä ļÆĘļ░øņ╣©ĒĢ£ļŗż[69]. Methionine/choline deficient dietļĪ£ ņ£ĀļÅäļÉ£ ļÅÖļ¼╝ ņŚ░ĻĄ¼ņŚÉņä£ CHOP deficiencyļŖö ņ¦Ćļ░®Ļ░äņŚ╝, Ļ░äņä¼ņ£ĀĒÖö, Ļ░äņĢöņØś ļ░£ņāØņØä ņÖäĒÖöņŗ£ĒéżļŖö Ļ▓░Ļ│╝ļź╝ ļ│┤ņŚ¼ņŻ╝ņ¢┤ CHOPņØś ņ¢ĄņĀ£Ļ░Ć ļŗżņ¢æĒĢ£ ņĢĮļ¼╝ ļ░Å ņāØļ”¼ņĀü ņåÉņāüņŚÉ ļīĆĒĢśņŚ¼ ļ│┤ĒśĖņĀüņØĖ ņŚŁĒĢĀņØä ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ│╝ ņłś ņ׳ļŗż[70-72]. Ļ▓īļŗżĻ░Ć CHOP Ļ│╝ļ░£ĒśäņØ┤ ņØĖĻ░ä ļ░Å ļ¦łņÜ░ņŖż Ļ░äņäĖĒżņĢö ļ¬©ļŹĖņŚÉņä£ Ļ┤Ćņ░░ļÉśņ¢┤ CHOPņØ┤ ņŚ╝ņ”Ø, ņä¼ņ£ĀĒÖö ļ░Å ņäĖĒż ņé¼ļ®ĖņØä ņ┤ēņ¦äĒĢ©ņ£╝ļĪ£ņŹ© Ļ░äļé┤ ļ░£ņĢö ļ░£ņāØņŚÉ ņŚ░Ļ┤ĆņØ┤ ņ׳ņØīņØä ļ│┤ņŚ¼ņżĆļŗż[73]. ĻĘĖ ņÖĖņŚÉļÅä ROS ļ░Å free radicalņØś Ļ│╝ņāØņé░ņ£╝ļĪ£ ņØĖĒĢ£ ņé░ĒÖöņŖżĒŖĖļĀłņŖżĻ░Ć ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ļ░£ņāØ ņøÉņØĖ ņżæ ĒĢśļéśņØĖļŹ░, ņ¦Ćļ░®ņ”ØņØ┤ ļ░£ņāØĒĢśļ®┤ņä£ ņ¦Ćļ░®ņé░ ĒŖ╣Ē׳ toxic lipidņØś ņČĢņĀüĻ│╝ ņ╣╝ņŖśņØś Ļ│╝ļČĆĒĢśĻ░Ć ļ░£ņāØĒĢśĻ│Ā ņØ┤ļŖö ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļź╝ ņ£Āļ░£ĒĢśļ®┤ņä£ ļ»ĖĒåĀņĮśļō£ļ”¼ņĢäņØś ĻĖ░ļŖźņØä ļ│ĆĒÖöņŗ£ņ╝£ ROS ļ░Å free radicalņØä Ļ│╝ņāØņé░ņŗ£Ēé©ļŗż. ĻĘĖļלņä£ toxic lipid peroxidation ļ░Å oxidative stressļŖö ņ¦Ćļ░®ņ”ØņŚÉņä£ ņ¦Ćļ░®Ļ░äņŚ╝ņ£╝ļĪ£ ņ¦äĒ¢ēņØä ņ£Āļ░£ĒĢśĻ▓ī ļÉ£ļŗż. ĻĘĖļ¤¼ļ»ĆļĪ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ņżæņÜöĒĢ£ Ļ░äņäĖĒż ņé¼ļ®ĖņØś ņżæņÜöĒĢ£ ĒŖĖļ”¼Ļ▒░ņØ┤ļ®┤ņä£, ņ¦Ćļ░®Ļ░äņŚ╝ņŚÉņä£ ņä¼ņ£ĀĒÖö ļ░Å Ļ░äņĢöņ£╝ļĪ£ņØś ņ¦äĒ¢ēņØä ņ£ĀļÅäĒĢśļŖö ņŚ╝ņ”ØņØś ņ×Āņ×¼ņĀü Ļ░ĆņåŹņØĖņ×ÉļĪ£ ņāØĻ░üĒĢ┤ ļ│╝ ņłś ņ׳ļŗż.
Ļ▓░ ļĪĀ
ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØĆ ļīĆņé¼, ņ£ĀņĀä ļ░Å ĒÖśĻ▓Į ņÜöņØĖņØä ĒżĒĢ©ĒĢ£ ņłśļ¦ÄņØĆ ĻĖ░ņĀäņŚÉ ņØśĒĢ┤ ļ░£ņāØļÉśļŖö ļ│Ąņ×ĪĒĢ£ ņ¦łļ│æņØ┤ļŗż. ņ¦Ćļ░®ņ”ØņØĆ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ĻĘ╝ļ│ĖņĀüņØĖ ĒŖ╣ņ¦Ģņ£╝ļĪ£ ļŗ©ņł£ ņ¦Ćļ░®ņ”ØņŚÉņä£ ņŚ╝ņ”ØĻ│╝ ņ¦äĒ¢ēņä▒ ņåÉņāüņØä ņ£Āļ░£ĒĢśļŖö ĒŖ╣ņĀĢ ņŗĀĒśĖ ļ®öņ╗żļŗłņ”śņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ļōżņØ┤ ņ¦äĒ¢ē ņżæņØ┤ļŗż. ņĄ£ĻĘ╝ ņŚ░ĻĄ¼ļōżņØĆ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżņÖĆ ļ╣äņĢīņĮöņś¼ņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśņØś ļ░£ļ│æ ļ░Å ņ¦äĒ¢ēĻ│╝ Ļ┤ĆļĀ©ļÉśņ¢┤ ņ׳ļŗżĻ│Ā ļ│┤Ļ│ĀĒĢśĻ│Ā ņ׳ņ£╝ļ®░, Ļ░äļé┤ ņ¦Ćņ¦ł ņČĢņĀüņØä ĒåĄĒĢ┤ ņ£Āļ░£ļÉ£ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļŖö ļ»ĖņĀæĒĢ® ļŗ©ļ░▒ņ¦ł ļ░śņØæņØä ĒÖ£ņä▒ĒÖöĒĢśņŚ¼ ņŚ░Ļ▓░ļÉ£ ņŗĀĒśĖņ▓┤Ļ│äļź╝ ĒåĄĒĢ┤ ļŗ©ņł£ ņ¦Ćļ░®ņ”ØņŚÉņä£ ņ¦äĒ¢ēņä▒ ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśĻ│╝ ņŚ░Ļ┤ĆļÉśņ¢┤ ņ׳ļŗżĻ│Ā ļ│┤Ļ│ĀĒĢśĻ│Ā ņ׳ļŗż. ņØ┤ļ¤¼ĒĢ£ ņŚ░ĻĄ¼Ļ▓░Ļ│╝ļōżņØĆ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖżļź╝ ņĪ░ņĀłĒĢśļŖö Ļ▓āņØ┤ ņ¦Ćļ░®Ļ░äņØś ļ░£ņāØņØä ņśłļ░®ĒĢśĻ│Ā ņ╣śļŻīļź╝ ĒĢśļŖö ļŹ░ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢĀ ņłś ņ׳ņØä Ļ▓āņ×äņØä ņĀ£ņŗ£ĒĢśĻ│Ā ņ׳ļŗż. ĻĘĖļ¤¼ļéś ņĢäņ¦üĻ╣īņ¦Ć ņ¦Ćļ░®Ļ░ä ņ¦łĒÖśĻ│╝ ņåīĒżņ▓┤ ņŖżĒŖĖļĀłņŖż ļ░śņØæ ĻĖ░ņĀäņŚÉ ļīĆĒĢ┤ņä£ļŖö ļ│┤ļŗż ļŹö ņ×ÉņäĖĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśļ®░, ĒŖ╣Ē׳ ņŗżņĀ£ ņ×äņāüņŚÉņä£ ņ¢┤ļ¢╗Ļ▓ī ņĀüņÜ®ļÉĀ ņłś ņ׳ļŖöņ¦ĆņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ļČĆņĪ▒ĒĢ£ ņāüĒā£ņØ┤ļ»ĆļĪ£ ņØ┤ņŚÉ ļīĆĒĢ£ ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśĻ▓Āļŗż.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






