


INTRODUCTION
Post-transplant lymphoproliferative disorder (PTLD) is a spectrum of clinically and morphologically heterogeneous lymphoid proliferations of various clonal compositions that are seen after bone marrow (BM) or solid organ transplantation (SOT) [1]. Although PTLDs represent rare complications (< 1%) after allogeneic hematopoietic stem cell transplantation (HSCT), known risk factors include donor/recipient human leukocyte antigen (HLA) mismatch, T cell-depleted hematopoietic stem cell grafts, use of anti-thymocyte globulin (ATG), and graft-versus-host disease (GVHD) [2]. The reported incidence for patients with these risk factors is as high as 15-25% [1,2]. The pathogenesis of PTLD in most patients appears to be related to B-cell proliferation induced by Epstein-Barr virus (EBV) infection. In primary or secondary immunodeficiency, acute EBV infection leads to a polyclonal expansion of B cells; these latently infected B cells may undergo uncontrolled proliferation and transformation into EBV-associated B cell lymphoproliferative disorders such as PTLD, which is a life-threatening complication [3]. In patients who undergo HSCT, EBV-associated PTLD is classified according to the time of onset: in the early post-transplantation period (< 1 year) or as a late manifestation (> 1 year) [3]. Early-onset PTLDs are usually EBV-positive, histologically heterogeneous, include most polyclonal mononucleosis-like forms, and are associated with higher peripheral blood (PB) EBV DNA levels [4]. Extranodal involvement is common in PTLD, whereas PB and BM involvement is extremely rare, with only a few reports of BM PTLD in recipients of BM and solid organ transplants [5,6]. Here, we describe the case of a 38-year-old woman with early-onset EBV-associated PTLD in PB and BM after haploidentical HSCT for Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL).
CASE REPORT
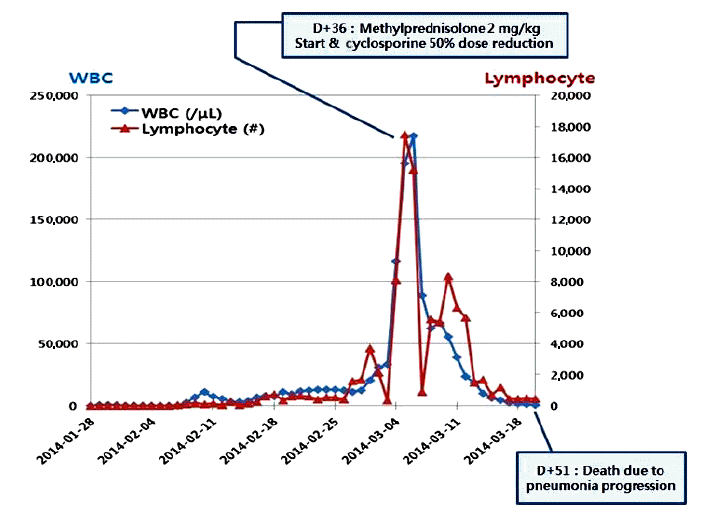
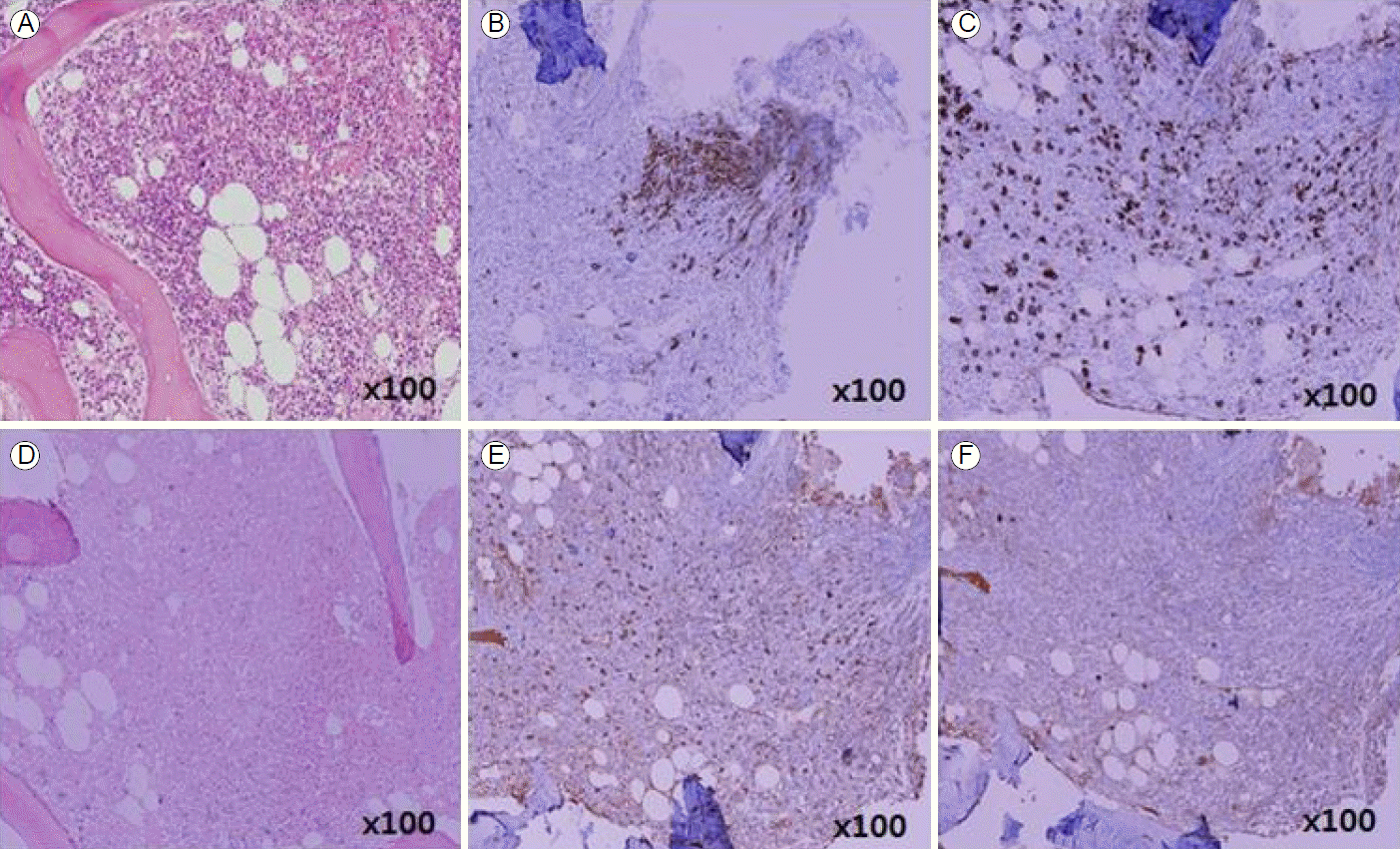
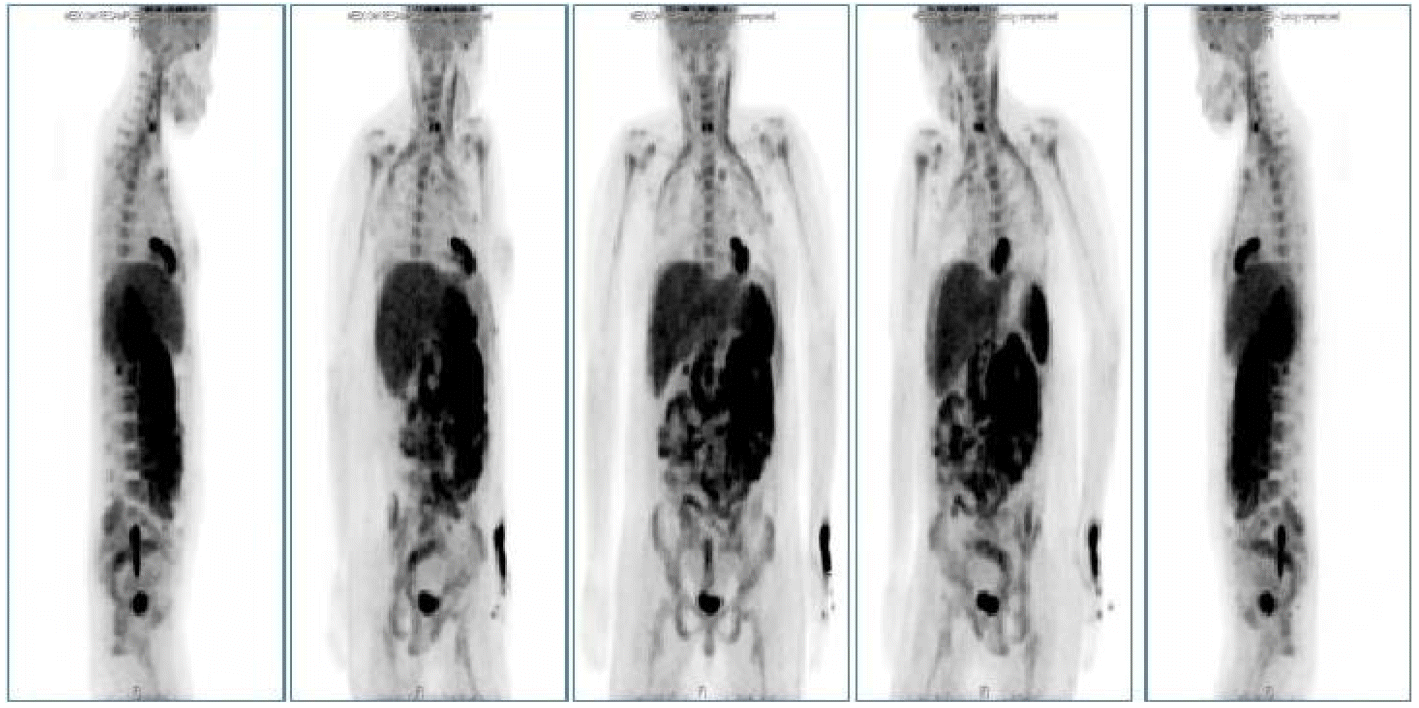
A 38-year-old woman was diagnosed with Ph+ ALL in October 2013. A BM chromosome study gave uninterpretable results and fluorescence in situ hybridization revealed t (9;22) and de l (9p21). Hematologic complete remission was achieved after the first remission induction chemotherapy using hyperfractionated cyclophosphamide, vincristine, doxorubicin, and dexamethasone with imatinib. The patient successfully received a haploidentical HSCT from her mother in January 2014. Non-myeloablative conditioning, consisting of fludarabine (30 mg/m2/day on days -7 to -2), busulfan (3.0 mg/kg/day on days -7 and -6), and ATG (2.5 mg/kg/day on days -4 to -1), was used as a preparative regimen. Standard GVHD prophylaxis, consisting of cyclosporine and short-course methotrexate on days +1, +3, +6, and +11 was given. On days 0 and 1, a granulocyte colony stimulating factor-mobilized peripheral graft with 6.08 ├Ś 106 CD34+ cells/kg and 2.62 ├Ś 108 CD3+ T cells/kg was transplanted. The times to recover to an absolute neutrophil count > 0.5 ├Ś 109/L, and a platelet count > 20 ├Ś 109/L without transfusion (reported as the first of 3 consecutive days), were 11 and 8 days, respectively. On day 14, a BM study revealed morphologically stable engraftment with three-lineage maturation, and chimerism analysis showed complete donor chimerism (CC). EBV serology before transplantation revealed IgG-positive (46.7) EBV-viral capsid antigen (VCA) and IgM-negative (< 10.0) EBV-VCA, indicating past infection. The donorŌĆÖs EBV serology before peripheral graft mobilization was EBV-VCA IgG positive (692.0) and EBV-VCA IgM negative (< 10.0). The similar pre-transplant EBV status of the donor and recipient suggested that the EBV infection was not primary, but chronic. On day 32, while receiving cyclosporine, the PB lymphocyte count increased suddenly to 3.7 ├Ś 103/╬╝L and continued to increase to 15.2 ├Ś 103/╬╝L on day 37 (Fig. 1). A BM study, chimerism analysis, and real-time quantitative polymerase chain reaction (PCR) for Bcr-Abl were performed on day 34. There was no evidence of leukemic relapse or loss of CC, except for polymorphic proliferation of a B-cell lineage expressing EBV latency-associated RNA in the BM. Immunohistochemical staining of the BM showed CD20-positive polymorphic lymphoid cells and CD138-positive plasma cells with lambda light chain restriction. In situ hybridization for EBV-encoded small nuclear RNAs mRNA demonstrated a few cells expressing nuclear signals in the BM (Fig. 2). Cytoplasmic immunoglobulin analysis of the PB mononuclear cells showed CD38-positive, CD138-positive, and CD19-positive cells with a lambda light chain restricted pattern, suggesting the presence of clonal plasmacytosis. Quantitative PCR for EBV-DNA in the PB showed that levels had increased to as high as 38,825,576 copies/mL on day 39. Whole-body 18F-fluorodeoxyglucose positron emission tomography (FDG-PET) on day 37 showed FDG uptake in the spleen, duodenum, jejunum, and multiple lymph nodes (mesentery and retroperitoneum). The PET findings did not indicate leukemic infiltration, but a reactive change due to inflammatory conditions (Fig. 3). These findings were consistent with a diagnosis of early polymorphic-type EBV-associated PTLD. The patient was managed by reducing the cyclosporine dose and administering high-dose steroid. On day 38, pneumonia and septic shock developed suddenly and worsened gradually. In the early phase of HSCT, although the absolute neutrophil count recovered, cellular immunity was still altered. Abnormal lymphocytosis due to PTLD also interferes with normal immune reconstitution. In addition, cyclosporine and high-dose steroid render patients vulnerable to acquired opportunistic infections. Unfortunately, our patient did not recover from the pneumonia and severe septic shock, which caused other organ damage, including cholestatic hepatitis and acute renal failure. She ultimately died of pneumonia with multiorgan failure on day 51, despite intensive treatment.
DISCUSSION
Epstein-Barr virus-associated PTLD is a well-documented complication in primary or secondary immunocompromised patients. The incidence of EBV-associated PTLD after HSCT is generally < 1%, but intensive immunosuppression, as in HLA-mismatched and T cell-depleted recipients, may markedly increase the risk of PTLD [1]. Park et al. [7] investigated the incidence and clinical progress of PTLD after allogeneic HSCT. The overall incidence of PTLD was 0.6% and there were no postmortem diagnoses. The overall response rate was 71.4% and the mortality rate was 42.8%.
Factors associated with an increased risk of PTLD include T-cell depletion and donor/recipient HLA mismatching. Even in the absence of T-cell depletion, the use of intensive immunosuppressive prophylaxis or GVHD therapy, especially anti-T cell agents such as ATG or OKT3, has been associated with the development of PTLD. Cellular immunity, as measured by the cytotoxic T lymphocyte response against EBV, does not re-develop until approximately 6 months post-HSCT; most cases of PTLD develop during this period [3]. The role of HLA mismatching in the pathogenesis of PTLD is unclear; it has been hypothesized that a mismatched graft is a source of chronic antigenic stimulation or delayed immune reconstitution. HLA-matched sibling HSCT is reported to have a relatively low incidence of EBV lymphoproliferative disorder (LPD). Recently, Gross et al. [8] estimated a 15.7% risk of LPD in HLA-mismatched and T celldepleted recipients at 6 months after transplant and suggested that older donor age was an independent risk factor for PTLD. Reddiconto et al. [9] investigated the incidence of EBV-related complications in 335 patients undergoing HSCT and documented an overall rate of 4.5%, with an incidence of 3.3% in myeloablative and 7% in non-myeloablative conditioning, suggesting a significantly higher risk of EBV-related complications in recipients of non-myeloablative conditioning. Therefore, although the exact etiology of PTLD remains unclear, it is believed to be multifactorial and probably involves impaired immunosurveillance of neoplastic cells, as well as depressed antiviral immune activity. In our case, because there was no HLA-matched sibling or unrelated donor, the haploidentical maternal donor was chosen as a source of stem cells for HSCT. Therefore, donor/recipient HLA mismatching and a non-myeloablative preparation regimen with ATG are risk factors for PTLD.
The origin of EBV in PTLD varies. In SOT, the PTLD cells are typically of recipient origin, suggesting that EBV infection represents reactivation of previously quiescent virus. Most cases of PTLD in allogeneic HSCT involve seropositive donors and recipients. LPD is believed to be donor-derived because the host lymphoid system has been eradicated by the conditioning chemotherapy or total body irradiation. Even in situations where the donor is seronegative, cases of PTLD have been shown to be of donor origin, suggesting that donor cells were infected with EBV after the transplant. In the current case, the pre-transplant donor EBV status was EBV-VCA IgG-positive and EBV-VCA IgM-negative, indicating past infection. Furthermore, a study of recipients of T cell-depleted grafts suggested that an elevated EBV-DNA load was highly predictive of the development of EBV-associated LPD. In our case, post-transplant lymphocyte count elevation, followed by a quantitative increase in EBV PCR to 38,825,576 copies/mL on day 39, was observed. These findings suggest that donor-derived EBV-associated PTLD had developed. Therefore, surveillance techniques using EBV antibody titers or EBV quantitative PCR should be checked before allogeneic HSCT in both donors and recipients to determine the origin of EBV infection. Monthly EBV PCR for circulating EBV DNA may be appropriate after allogeneic HSCT, particularly in high-risk settings such as T-cell depletion, HLA-mismatched HSCT and EBV-seromismatched SOT. Jin et al. [10] reported two cases of HodgkinŌĆÖs lymphoma after allogeneic HSCT for acute myeloid leukemia. One of these patients received HLA-mismatched unrelated HSCT and developed EBV-positive HodgkinŌĆÖs lymphoma 36 months after the transplant. The patient continued taking mycophenolate mofetil because of a history of acute GVHD. Taken together with our case, an HLA-mismatched donor, prolonged immunosuppression treatment, and EBV infection are important risk factors for PTLD development.
Extranodal involvement is common in PTLD, whereas PB and BM involvement is extremely rare. There are only a few reports of BM PTLD in SOT recipients. Cho et al. [6] reported BM involvement in 4 of 23 (17.4%) patients with PTLD, two of whom had BM lesions without involvement at other sites. To the best of our knowledge, ours is the first case report describing PB and BM involvement of PTLD after allogeneic HSCT for Ph+ ALL.
Previous studies have reported using treatments such as immunosuppression reduction, cytotoxic chemotherapy, monoclonal antibody therapy, radiation therapy, cellular immunotherapy, antiviral therapy, and cytokine therapy [2]. A massive reduction of immunosuppression represents the most important treatment. Recent results with anti-B-cell monoclonal antibody therapy, in conjunction with rituximab and cytotoxic T lymphocyte therapy, have been quite promising, with many patients achieving long-term relapse-free survival [2]. In lymphoma with positive EBV markers, antiviral therapy with acyclovir or ganciclovir can be considered. However, although acyclovir and ganciclovir inhibit viral DNA polymerase, they have no effect on cellular DNA polymerase and do not inhibit in vitro EBV-induced transformation. Consequently, there is no obvious role for acyclovir or ganciclovir as either prophylaxis or treatment for PTLD. In our patient, we reduced immunosuppression and administered steroid, which proved effective against PTLD.
Although PTLD is rare during the early post-transplantation period, we should consider the possibility of PTLD when encountering early peripheral lymphocytosis with no clear cause. Since there is no method for predicting PTLD, and PTLD in HSCT tends to progress rapidly, it is often fatal relatively early after transplantation; an early diagnosis of PTLD is important for prompt therapy and to decrease mortality. Some studies have reported that the early mortality rate for PTLD after HSCT approached 90% [2]. Therefore, if there is abnormal peripheral lymphocyte proliferation in the early phase of allogeneic HSCT, an EBV activity test (EBV quantitative PCR, EBV-VCA or EBV nuclear antigen), lymph node biopsy, and FDG-PET should be performed. When PTLD is confirmed, the immunosuppressive dose should be reduced first, which often eliminates the disease permanently. Additional therapies should be tailored to the individual patient and clinical situation.
In conclusion, PTLDs are fascinating disorders that clearly highlight the roles of the immune system and viral infection in the development of malignancy. The pathogenesis of EBV-associated PTLD is thought to be multifactorial and is not well understood. Therefore, supportive care and adopted immunotherapy should be integrated into the treatment regimen of this aggressive and often fatal disease. Based on our experience, further long-term studies should examine patients with early peripheral lymphocytosis and EBV-associated PTLD after allogeneic HSCT. Moreover, guidelines should be prepared that enable the prediction of this complication after haploidentical HSCT.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






