


서 론
혈관 석회화는 혈관 내부 조직에 칼슘-인산염 형태의 병리적 침착을 말한다. 이는 정상 노화 과정에서 발생하기도 하지만 당뇨병, 심혈관질환 및 유전질환 등의 특정 질병 상태에서 흔하며 그 진행이 가속되기도 한다. 혈관 석회화의 가장 흔한 원인 질환 중 하나가 만성 콩팥병이다. 성인 말기 신부전 또는 투석 환자에서는 혈관 석회화가 매우 흔하며, 또한 소아 투석 환자 및 초기 단계 만성 콩팥병 동물 모델에서도 혈관 석회화 발생 및 악화를 보고하기도 하여 그 중요성을 주목할 만하다[1,2].
혈관 석회화는 일단 발생하게 되면 심혈관질환으로의 이환 및 사망률의 증가와 밀접한 관계를 갖는데, 혈관벽 탄력 소실이 맥압 상승을 유발하고, 궁극적으로 좌심실 비대를 초래하여 심혈관질환 합병증 가능성을 높이는 것으로 설명되고 있다[3].
혈관 석회화는 혈관벽의 내막 석회화, 중막 석회화, 심장판막 석회화 및 저항성 칼슘 형성(calciphylaxis, calcific uremic arteriolopathy)으로 구분할 수 있다. 이 중 내막 석회화는 이상지질혈증, 염증 및 내막 비후와 연관되어 죽상동맥경화증을 초래하는데, 내부 혈관벽에 불안정하고 파열될 수 있는 플라크를 형성하여 폐쇄성 혈관질환을 유발한다. 이에 비하여, 중막 석회화(Mönckeberg’s arteriosclerosis)는 주로 당뇨병 또는 만성 콩팥병에서 흔하다. 혈관내강 협착이 없이도 발생할 수 있으므로, 주로 혈관 경직화를 유발하여 심혈관질환 합병증 발생을 높인다. 그러므로 이러한 혈관 석회화에 관한 보다 깊은 이해가 만성 콩팥병 환자의 합병증을 최소화하는 최적의 치료법을 발전시키는데 필수적일 것이나, 아직까지 명확한 답을 제시하는 것은 불가능할 것으로 생각된다. 이에 본 종설에서는 만성 콩팥병 환자에서 특징적인 중막 석회화에 관한 기전을 최근의 연구를 중심으로 살펴보고, 진단 및 치료에 관한 현재까지의 연구 결과 및 임상 적용에 관하여 살펴보고자 한다.
본 론
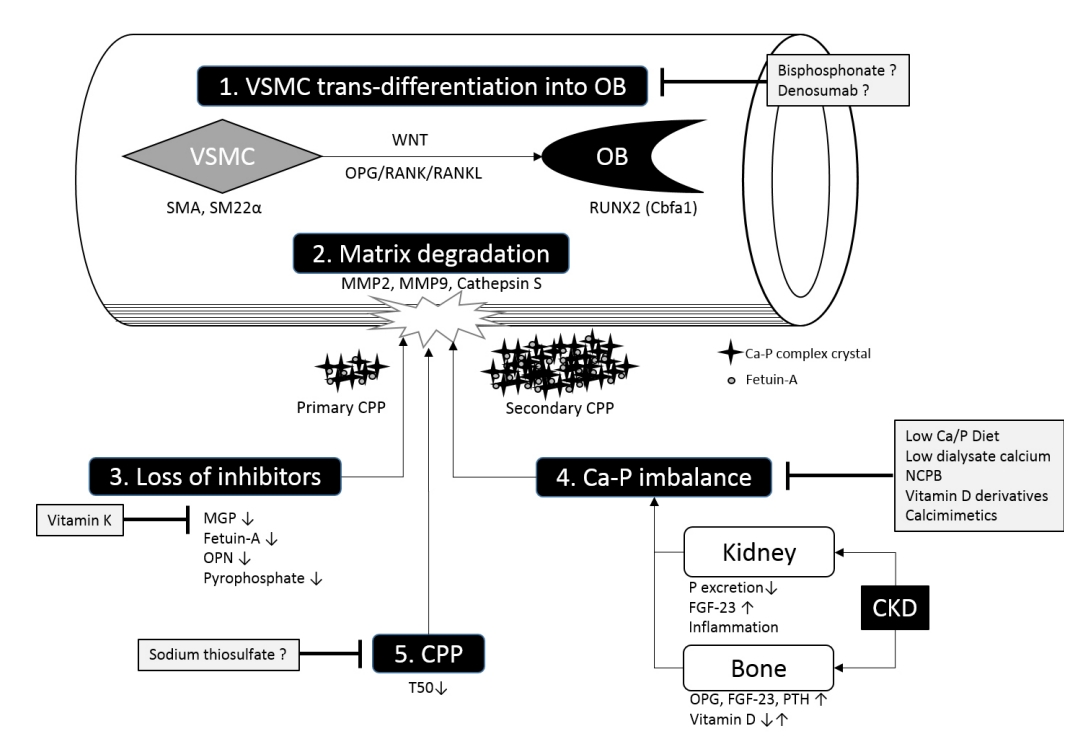
혈관 석회화의 병태생리(pathophysiology of vascular calcification, Fig. 1)
혈관평활근세포의 변화 분화(trans-differentiation of vascular smooth muscle cells into osteoblast like cell)
혈관평활근세포가 변화 분화를 통하여 골아세포로 표현형 변화를 일으키는 것이 혈관 석회화 과정 중 가장 중요하고 필수적인 단계로 여겨진다[4,5]. Runt related transcription factor 2 (RUNX2 or core-binding factor α 1, Cbfa1)는 정상적인 골아세포 분화에 관여하는 주요 전사인자로서, 이러한 혈관평활근세포 표현형 변화에 가장 중요한 요소이다[6]. 혈관벽에 칼슘 부산물이 침착되기 전에 이미 혈관평활근세포 표현형 변화는 시작된다. 이는 골형성 세포와 유사한 형태를 보이는데, 혈관평활근세포의 특징적 유전자인 smooth muscle α-actin (SMA)와 smooth muscle protein 22α (SM22α)의 발현이 감소되고, 골연골화의 표지자인 RUNX2 (Cbfa1), osterix, osteopontin, osteocalcin, alkaline phosphate 등의 발현이 증가된다[4,5,7,8]. 이와 같은 골아세포 유사세포는 혈관평활근세포의 특징인 수축력을 소실하고 연골 기질 및 칼슘-인이 풍부한 기질 소포(matrix vesicle)를 형성하게 되어 혈관벽의 석화 작용이 촉발된다[4,5,9].
하지만 골형성과 혈관 석회화는 유사한 생리적 기전을 공유함에도 불구하고, 만성 콩팥병 환자에서 골감소성 골질환 및 혈관 석회화가 동시에 발생하는 ‘석회화 역설’은 현재까지도 연구자들에게 난제로 남아 있다[10]. 최근 골대사 및 골다공증에 관한 연구의 발전으로 인하여 osteoprotegerin (OPG)/receptor activator of NF-κB (RANK)/RANK ligand (RANKL) 신호 체계를 통하여 병태 생리에 관한 이해도가 높아지고 있다. 골재형성(bone remodeling) 또는 골회전율(bone turnover)은 손상된 골조직을 파골세포가 흡수하고 이를 골아세포가 재생성하는 과정의 반복을 의미하는데, OPG/RANK/RANK 시스템이 골재형성 조절에 중요한 역할을 한다. RANK는 파골세포 표면에 발현하는 수용체이며, 그 리간드가 RANKL로 이는 골아세포의 표면에 발현한다. 골조직의 손상에 반응하여 골아세포가 파골세포를 소환하게 되는데 이 때 RANKL/RANK 결합이 필요하다. 파골세포가 일단 골흡수 임무를 마치면 OPG가 골아세포 표면의 RANKL와 결합하여 더 이상의 파골세포의 소환 및 분화를 억제하는 유인 장치 역할을 수행한다. 골재형성 과정에 보다 많은 관심이 있는 독자들은 복잡한 기전을 이해하기 쉽게 애니메이션으로 소개한 Susan Ott (University of Washington) 교수의 웹주소를 소개하니 참고하기 바란다(http://courses.washington.edu/bonephys/Gallery/BMURemodel.swf). OPG는 골아세포 및 다양한 세포에서 분비되는데, 특히 혈관벽에서 분비되어 혈관 석회화를 저해하는 것으로 보고되고 있다[10]. OPG 제거 생쥐를 이용한 연구에서는 혈관 석회화 및 골다공증이 발생되었고[11], 혈관평활근세포에 RANKL을 투여하면 혈관 석회화 과정을 자극하는 결과를 보여[12], OPG와 RANKL은 각각 혈관 석회화의 발생의 억제제와 자극제로서의 역할을 수행하는 것으로 이해된다. 그러나 혈액투석 환자를 대상으로 한 연구에서는 OPG 농도가 오히려 높았고 혈관 석회화 정도와 비례하는 결과를 보여[13], 그 역할에 관해서는 추가 연구가 필요하겠다.
또 혈관 석회화와 연관된 또 다른 신호 전달 체계인 WNT 전달로에 대한 연구자들의 관심이 늘어나고 있다. Muscle segment homeobox protein homolog 2 (Msx2)가 WNT 전달로 활성으로 혈관 석회화를 유발한다고 보고된다[14]. WNT 전달로는 정형(canonical) 또는 비정형(non-canonical) 신호 체계로 구분되어지고, 이에 관한 골 대사 및 혈관 석회화에 관한 연구가 진행되고 있으나 LRP5/6, Wnt5a, Wnt5b, Wnt11 등과 연관성 연구 결과가 보고될 뿐 아직까지 병태생리를 설명할 수 있는 특정 하부 신호체계 및 치료 목표 물질이 발견되지는 않았다[15,16]. 또한 정형 WNT 전달로 활성화는 골아세포 활성에 관여하며, 최근 골다공증에 관한 이해도가 증가하고 새로운 치료제가 개발됨에 따라, 이에 관한 연구는 앞으로 활발해질 것으로 예상된다[17,18].
혈관세포외기질 소실(matrix degradation)
Elastin은 혈관벽에 가장 풍부하게 존재하는 단백 물질로, 만성 콩팥병 환경에서는 elastin의 소실이 흔하다[19,20]. Elastin 소실은 matrix metalloprotease (MMP)로 대표되는 단백분해효소 중 특히 MMP2와 MMP9에 의한 것으로 생각되는데, 만성 콩팥병 환경은 MMP의 발현이 증가된 상태이므로 MMP와 elastin이 결합하게 되면 결과적으로 elastin의 소실을 초래한다[19,20]. 흥미로운 점은 이전 동물 실험연구에서 신기능 감소로 유발된 elastin 소실만으로는 혈관 석회화를 유발하지 못하고, 고인산 식이를 통한 고인산혈증이 혈관 석회화 유도에 필수적이라는 점이다[19]. 연구자들은 미네랄 조절 장애 환경에서 elastin이 붕괴된 부분이 칼슘 침착이 용이하도록 침전 장소를 마련하는 과정으로 생각된다고 주장하였다[4,19]. 또한, 아포리포단백질 E 제거 생쥐를 이용하여 만성 콩팥병을 유도한 동물 연구에서는 단백효소인 cathepsin S의 상승이 elastin 소실에 필수적임을 보고하기도 하였다[21].
석회화 억제 기전의 소실(loss of anti-calcific mechanisms)
정상적인 환경에서는 광범위한 조직 석회화를 방지하기 위하여 미네랄 형성을 억제하는 다수의 조절 물질이 존재하는데, 이는 혈관 석회화 억제제로서의 역할을 수행한다. 만성 콩팥병 환자에서는 이러한 억제제의 기능이 비정상적이므로 석회화 성향을 보일 뿐만 아니라 기존 석회화 부분도 악화된다[22].
Matrix Gla protein (MGP)은 뼈, 연골, 콩팥, 폐, 심장 또는 혈관평활근에 존재하는데, 칼슘 이온과 결합하여 잉여 칼슘을 제거하며 칼슘 결정과도 결합하여, 결과적으로 결정체 성장을 억제하는 역할을 한다[23,24]. 만성 콩팥병 환자에서 고인산혈증은 MGP와 칼슘 이온의 결합을 약화시키고, 비타민 D 결핍은 MGP 발현을 감소시킴으로써, 결과적으로 석회화 경향을 보이게 된다[25]. MGP의 활성화를 위해서는 카르복시화(γ-carboxylation) 과정이 필요하며, 비타민 K가 보조인자로 작용한다[24,26]. 그러므로 만성 콩팥병 환자에서 흔히 사용되는 항응고제인 와파린은 MGP 활성화 과정을 억제한다. 이는 혈액투석 환자에서 와파린 투여와 연관된 혈관 석회화 악화 및 비타민 K 투여를 통한 혈관 석회화 완화를 설명하는 기전이 된다[27-29]. 더불어, 말기신부전 환자에서 혈청 비활성 MGP (dephosphorylated uncarboxylated MGP)의 상승이 혈관 석회화 및 심혈관계 질환 합병과 연관된 사망의 주요 예측인자로서의 가치를 보고하기도 하였다[30-32].
또 다른 중요한 억제제로 간에서 합성되는 당단백인 fetuin-A (α2 Heremans-Schmid glycoprotein, AHSG)가 있다. 이는 정상 상태에서 혈액 내에 높은 농도로 존재하고, 주로 칼슘 이온과 수산화인회석(hydroxyapatite)과 결합하여 혈관평활근세포의 사멸을 감소시키고, 소포내 인산칼슘결정 형성을 방해한다[33]. Fetuin-A 제거 생쥐를 이용한 연구에서 광범위한 석회화가 확인되었고[34], 혈관평활근세포에 fetuin-A를 추가하여 석회화 경향의 감소를 유도한 생체외 연구 결과를 보고하기도 하였다[22]. 혈액투석 환자에서 혈청 fetuin-A의 감소와 혈관 석회화와의 역 상관관계 및 사망위험도와의 양의 상관관계를 보고한 외국의 여러 연구와 더불어[35-37], 국내 연구자들도 복막투석 환자에서 fetuin-A 감소와 연관된 단일염기다형성 및 혈관 석회화와의 연관성을 보고한 바 있다[38]. 그러나 투석 전 당뇨병 환자를 대상으로 한 연구 또는 투석 환자를 대상으로 추적 관찰 연구에서는 fetuin-A와 혈관 석회화와의 역 상관관계를 보이지 않음을 보고하여[39-41], 이러한 결과의 차이를 설명하기 위해서는 fetuin-A 외 별도의 설명 요소가 필요할 것으로 생각되며, 본 종설의 후반부에서 석회화 단백입자(calciprotein particle)를 통하여 추가로 살펴보고자 한다.
Osteopontin (OPN)은 인단백질로 정상적으로 뼈와 치아에 존재하며, 인회석 결정체(apatite crystal) 형성을 저해하고 파골세포를 활성화시킨다[42,43]. 일반적으로 혈관구조 내에서는 발견되지 않고 석회화된 혈관에서 발현이 증가되는 것으로 볼 때[44], 혈관 석회화 진행 과정에서 혈관이 훼손되거나 병변이 발생되면 OPN이 활성화되어 혈관 재형성을 수행하는 것으로 이해된다[4]. 만성 콩팥병 환자에서는 대조군에 비하여 OPN이 증가 소견을 보이나, 혈관 석회화와의 연관성에 다양한 결과를 보이는 것으로 보고되어, 혈관 석회화에서 OPN의 역할에 관해서는 추가적 연구가 필요한 상태이다.
Pyrophosphate는 혈관평활근에서 생성되어 혈장에 존재하며, 정상 상태에서 수산화인회석(hydroxyapatite) 형성을 방해하는 자연 억제제 중 하나이다. Pyrophosphate 생성에는 ectonucleotide pyrophosphate phosphodiesterase 1 (ENPP 1)이 필요하며, 상염색체 열성 유전 형태로 ENPP 1의 기능 소실 돌연변이가 특발성 신생아 혈관 석회화를 일으키는 것으로 보고되었다[45]. 동물 연구에서는 ENPP 1 제거 혈관을 야생형 동물 개체에 이식한 경우 혈관 석회화는 발현되지 않았으나, ENPP 1 제거 동물 개체에 야생형 혈관을 이식한 경우에는 혈관 석회화가 발현되었다[46]. Pyrophosphate는 콩팥을 통하여 제거되나 혈액투석 환자에서 오히려 낮은 농도를 보여[47,48], pyrophosphate가 투석을 통하여 제거되는 점을 감안하더라도 말기신부전 상태에서 pyrophosphate 생성 감소 또는 콩팥 외 대사 증가 등의 가능성에 관한 추가 연구가 필요할 것으로 생각된다.
칼슘 인 불균형(imbalance of calcium and phosphorus homeostatsis)
고칼슘 환경에서 혈관평활근세포는 골아세포 유사세포로 변화 분화를 일으키는데, 아마도 세포 내로 인산 이동의 증가가 원인이라 주장하는 실험실적 연구 결과가 보고된 바 있다[49]. 특히 L-type 칼슘 통로가 혈관 석회화와 연관되며, 통로 차단제인 verapamil 투여로 혈관평활근세포의 석화작용을 억제하는 결과가 보고되었다[50].
고인산혈증은 만성 콩팥병과 말기신부전 환자에서 흔히 관찰되는 소견으로 인산염의 콩팥 배설 감소가 그 원인이다. 혈관 석회화 과정에서 고인산혈증의 영향은 주로 나트륨 의존성 인산염 운반기(sodium-dependent phosphate transporter; NaPi)와 연관될 것으로 이해된다[51]. NaPi type I과 type II는 주로 콩팥과 장에 발현되어 인산염의 전신 항상성 유지 역할을 수행하는 반면, NaPi type III (inorganic phosphate transporter [PiT]-1, PiT-2)는 콩팥을 포함한 여러 장기 및 혈관평활근 및 골아세포에도 보편적으로 발현되어 주로 국지적인 인산염 조절에 관련하여 혈관 석회화에 영향을 미친다고 이해된다[52,53]. PiT-1 유전자 억제를 이용한 생체외 연구에서 PiT-1은 혈관평활근세포의 표현형 변화에 연관되며, PiT-1이 감소된 환경에서는 PiT-2 발현이 증가되어 보상적으로 작용함이 보고되기도 하여[52], 향후 추가적 연구가 우리의 이해를 도울 것으로 예상해 볼 수 있다.
만성 콩팥병 환자에서는 고인산혈증이 발생되기 전부터 이미 parathyroid hormone (PTH)과 fibroblast growth factor-23 (FGF-23)의 상승을 보이는 경우가 흔한데, 이는 PTH와 FGF-23가 인산뇨를 유발하는 호르몬으로서의 역할을 수행하기 때문이다. 혈액투석 환자에서 FGF-23의 증가와 혈관 석회화와의 연관성이 보고된 바 있으나[54], 대규모 코호트 연관성 연구(Chronic Renal Insufficiency Cohort [CRIC] Study) 및 혈관평활근세포 배양액에 FGF-23을 추가한 생체외 연구 결과에서는 혈관 석회화에 미치는 영향이 불확실한 것으로 보고하기도 하여[55] 추가적 연구가 필요할 것으로 생각된다. 또한 국내 만성 콩팥병 코호트 연구(KoreaN cohort study for Outcome in patients with CKD, KNOW-CKD study)에서는 정상 체질량지수를 보이더라도 중심성 비만을 보이는 환자의 경우가 혈관 석회화 위험도가 높은 것으로 분석되어[56], 단순히 미네랄 불균형뿐 아니라 인구 생태학적 요인도 영향을 미칠 수 있음을 시사하는 결과라 할 수 있겠다.
PTH 또한 만성 콩팥병 환자에서 칼슘 및 인 조절에 많은 역할을 수행하는 것으로 잘 알려져 있으며, 이차성부갑상선항진증의 조절 목적으로 투여하는 칼슘유사작용제(calcimimetics)가 혈관 석회화의 진행을 다소 감소시킨다는 임상연구 결과가 보고되기도 하였다[57]. FGF-23과 PTH는 칼슘 및 인과 더불어 만성 콩팥병 미네랄 골질환(chronic kidney disease-mineral bone disease, CKD-MBD)과 연관되어 질환 발생의 중요한 요소일 뿐 아니라 그 진단 및 치료의 탐지자로서의 기능도 알려지고 있는 바, 앞으로도 지속적인 연구자들의 관심분야가 될 것임을 예상할 수 있겠다.
석회화 단백 입자(calciprotein particle)
앞에서 언급된 fetuin-A는 1961년 Hermans, Schmid, Burgi 등에 의하여 인체 당단백으로 발견되어 단순히 혈관 석회화 억제제로서 작용하는 것으로 알려졌으나, 최근에는 단순히 칼슘과 결합하는 것이 아니라 fetuin-mineral complex, 즉 calciprotein particle (CPP)을 형성하여 혈관 석회화 발생 및 악화를 방해하는 작용을 하는 것으로 이해되고 있다. CPP는 두 단계를 거쳐 형성되는데, primary CPP가 먼저 형성된 후, 보다 크기가 크고 복잡한 구조로 육인산칼슘복합체(octacalcium phosphate complex)로 구성된 중심부에 고밀도의 fetuin-A가 주위를 둘러싸는 형태의 secondary CPP를 단계적으로 형성하게 된다[58-60]. CPP의 측정은 혈장의 단계적 원심분리를 통한 간접적 방법이 있으나[58], 최근에는 직접 CPP를 측정하는 방법이 개발되어 보고되었다[59]. 또한, primary CPP가 secondary CPP로 변화하는 시간을 나노입자 기반 측정법(nanoparticle-based assay)을 통하여 석회화 성향(calcification propensity)인 T50를 측정할 수 있는데[61], 혈액투석 환자에서 T50와 혈관 경직도와의 역 상관관계 및 사망률과의 연관성이 보고되었다[62,63]. CPP는 세망내피계로 제거되므로, 형성에서 작용 및 제거에 이르기까지 만성 콩팥병 환경에서 그 역할에 관한 추가적 연구가 필요하며, 향후 혈관 석회화 관련 연구에서는 기존 fetuin-A의 단순 측정보다 CPP 및 T50가 차지하는 부분이 점차 커질 것으로 예상된다.
혈관 석회화의 진단
혈관 석회화의 진단 및 추적에는 단순 일반 방사선 사진, 2차원 초음파 및 전자빔컴퓨터단층촬영(electron beam computed tomography, EBCT)이 이용된다.
EBCT는 Agatston score를 이용하여 석회화 영역의 조밀도와 수량 정도를 정확히 계산할 수 있다[64]. EBCT를 통한 혈관 석회화의 정량법은 심혈관질환 합병증을 예측하는데 유용한 것으로 보고되었다[65]. 2차원 초음파의 경우, 경동맥과 대퇴동맥과 같은 표재 동맥의 석회화 측정에 용이하고, 최근의 초음파 기계는 맥파 속도를 측정할 수 있는 기능이 있어 동맥 순응도 및 경직도를 함께 평가할 수 있는 장점이 있다. 그러나 초음파의 경우의 술자에 영향을 받을 수밖에 없고 반정량적 측정만 가능하며, EBCT와 초음파를 통한 측정법은 내막 석회화와 중막 석회화를 구분하기 어렵다는 단점이 있다[66].
더불어, 임상적으로 보다 접근이 용이한 단순 영상을 통한 혈관 석회화 정량법이 고안되었다. 흉부 방사선 촬영 및 측면 복부대동맥 촬영을 통한 복부대동맥의 혈관 석회화를 정량화 할 수 있다. 혈액투석 환자에서 흉부방사선 촬영을 이용한 혈관 석회화 정량법은 EBCT로 측정한 혈관 석회화와 비교하여 유효성이 입증되었고, 심혈관질환 합병 및 사망률과 연관성을 보였다[67,68]. Kauppila 등[69]에 의하여 고안된 측면 복부대동맥 촬영을 이용한 복부대동맥 석회화의 정량(0-24점)은 신기능이 상대적으로 양호한 환자에서 분석한 결과가 보고된 후, Bellasi 등[70]에 의하여 혈액투석 환자에서 측면 복부대동맥 촬영으로 정량한 혈관 석회화 점수가 7점 이상일 경우 EBCT로 측정된 혈관 석회화 점수가 100 이상일 가능도(likelihood)를 분석하여 그 유효성을 보고한 바 있다. 단순 방사선 촬영에서 내막 석회화는 불규칙하고 고르지 못한 형태를 보이는 반면, 중막 석회화는 균등한 선형 형태의 석회화를 보인다. 만성 콩팥병 및 말기신부전 환자에서는 중막 석회화가 흔하고 중요한 소견이며, 말초근육 주변 동맥에 흔하게 관찰되므로, Adragao 등[71]은 골반 및 수부 방사선 촬영을 통하여 혈관 석회화 정량법(0-8점)을 고안하였다. 장골, 대퇴골, 요골 및 수부 동맥의 혈관 석회화를 정량할 수 있으며, 혈액투석 환자에서 혈관 석회화 정도와 심혈관질환 합병증과의 연관성이 보고되었다[71].
현재까지, 만성 콩팥병 환자에서 혈관 석회화와 심혈관질환 합병증과의 연관성은 강력한 것으로 생각되고 있으나, 임상에서는 선별 검사 및 진단적 검사에 대한 관심과 노력은 부족한 실정이다. 혈관 석회화의 진행이 단기간 내 확인하기 어려울 뿐만 아니라, 적절한 치료법이 개발되지 않은 것이 주요한 원인이라 생각할 수 있겠다. The Kidney Disease Improving Global Outcome (KDIGO) work group에서는 2017년 임상 지침 업데이트에서 CKD-MBD와 연관지어 혈관 석회화의 진단에 EBCT를 표준 방법으로 소개하면서, 측면 복부대동맥 촬영을 이용하여 복부대동맥의 석회화를 정량하거나 심초음파를 통한 심장판막 석회화를 확인하는 것을 EBCT의 적절한 대안법으로 권장하고 있다(2C) [72]. 특히 진행된 만성 콩팥병 환자 및 투석 환자(CKD G3a-5D)의 경우 혈관 석회화를 동반하면 심혈관질환 위험도가 높을 것으로 생각하고(2A), CKD-MBD 치료에 혈관 석회화 정량법 활용을 권고하고 있으나, 적절한 치료법의 부재로 인하여 권장 정도가 낮은 편이다(not graded) [72].
혈관 석회화의 치료(Fig. 1)
최근까지도 혈관 석회화에 관한 적절한 치료 방법은 전무한 실정으로, 혈관 석회화의 위험요소를 제거하고, 미네랄 대사를 조정하거나, 혈관 석회화 진행을 다소 완화시킨다고 보고된 약제 처방이 최선이다.
칼슘 및 인 불균형 해소(calcium and phosphate balance)
만성 콩팥병 환자에서 대부분 저칼슘혈증을 보이나, 식이 섭취된 칼슘 및 투석액 칼슘 농도로 인하여 혈중 칼슘 농도에 영향을 미칠 수 있다. 현재까지의 공통된 의견으로는 가능한 낮은 혈중 칼슘 농도를 유지하는 것이 혈관 석회화 예방 및 완화를 위한 최선의 방법으로 생각되므로, 고칼슘 식이를 지양하고 혈액투석 환자의 경우는 저농도 칼슘 투석액(2.5-3.0 mEq/L; 1.25-1.50 mmol/L) 사용을 권장한다[72]. 하지만 초저농도 칼슘 투석액 사용이 임상적으로 유리하다는 증거는 미약하므로, 고칼슘혈증 치료 목적을 제외하고는 투석액 칼슘 농도를 2.5 mEq/L (1.25 mmol/L) 이하로 낮추는 것은 권장되지 않는다[73,74].
콩팥 기능이 감소함에 따라 요중 인산 배설이 감소되므로, 식사 중 인산 섭취가 증가한 경우는 고인산혈증을 보이게 된다. 식이 조절과 함께 요중 인 배설분획(fractional excretion of phosphate)을 측정하게 되면 인 섭취량 계산과 고인산혈증의 치료 계획을 세우는데 도움이 될 수 있는 것으로 알려져 왔으나, 최근에는 고인산혈증이 인 섭취량보다는 체내 주요 장기의 인산 흡수 및 배설률에 따라 일 중 변화를 보인다는 연구 보고가 계속되므로[75], 인산의 측정 시기 및 섭취량과의 연관성에 관해서는 추가적인 연구 및 의견 합의가 필요한 상황이다. 여하튼, 식이 조절로 인산 조절이 부적절한 경우는 인결합제 투여가 필수적이며, 비칼슘계 인결합제가 투석 환자에서 혈관 석회화의 진행을 완화시킨다는 연구 결과에 따라서[76,77], 최근에는 칼슘계 인결합제 사용은 가능한 지양하도록 권고하고 있다[72,76,77]. 많은 주요 연구에서는 비칼슘계 인결합제의 사용이 사망과 같은 주요 임상 결과에 이점은 보고되지 않았으나, 메타 분석에서는 비칼슘계 인결합제가 사망 위험도를 감소 시키는 것으로 분석되었다[77].
비타민 D 유도체(vitamin D derivatives)
활성 비타민 D 유도체의 사용은 주로 PTH 조절 목적이며, 투석 환자에서 사망률을 감소시킨다는 보고는 있으나[78,79], 혈관 석회화에 관한 개선 효과를 보이는 연구 결과는 부족하다. 동물 연구에서는 calcitriol을 초생리적 용량으로 투여하게 되면 혈관 석회화를 악화시키는 반면[80,81], 저용량의 calcitriol 또는 paricalcitol 투여는 혈관 석회화를 예방하는 것으로 보고되었다[82]. 혈액투석 환자에서는 alfacalcidol의 누적 투여량이 혈관 석회화와 양의 상관관계를 보였으며[83], 투석 전 만성 콩팥병 환자의 연구에서는 1,25-dihydroxy vitamin D와 혈관 석회화는 음의 상관관계를 보였다[84]. 이와 같은 결과로, 비타민 D의 생리적 용량 투여는 유해하지 않을 것으로 짐작할 수 있으나, 2017년 KDIGO CKD-MBD 임상 지침에서는 투석 전 만성 콩팥병 환자에서는 투여로 인한 이점이 명확하지 않고[85], 혈중 칼슘 농도 상승 위험성으로 인하여 활성 비타민 D 제제의 투여를 권장하지 않는다고 하였다[72].
비스포스포네이트(bisphosphonate)와 데노수맙(denosumab)
Bisphosphonate는 pyrophosphate 유사체로 파골세포 기능 및 골흡수를 방해한다. 혈액투석 환자에서 약 6개월 etidronate를 투여한 결과 혈관 석회화 완화를 보였다는 보고가 있으나[87,88], 투석 전 만성 콩팥병 환자를 대상으로 한 연구에서는 alendronate의 투여가 대조군에 비하여 혈관 석회화 완화에 미치는 영향이 미미함을 보였다[89]. 또한, 대규모 코호트 연구(The Multi-Ethnic Study of Atherosclerosis)에서는 bisphosphate 투여가 65세 이상의 고령 환자에서는 심혈관 석회화의 유병률을 감소시켰으나, 상대적으로 젊은 연령에서는 그 효과가 확실하지 않았다[90]. Bisphosphonate가 대부분 콩팥으로 배설되므로, 콩팥 기능이 감소된 환자에서 처방이 자유롭지 못하고, 골 침착 및 무력성 골질환의 위험성으로 인해 혈관 석회화의 치료제로서의 선택 가능성은 미미할 것으로 보인다.
Denosumab은 RANKL의 인간 단클론항체로 주로 간을 통하여 대사되므로, 콩팥 기능이 감소된 환자에서도 비교적 자유롭게 사용할 수 있는 새로운 골다공증 치료제이다. 혈관 석회화와 골형성의 유사성으로 인하여 denosumab의 투여가 혈관 석회화에 미치는 영향은 매우 흥미로운 부분이다. 동물 연구에서 denosumab 투여로 인하여 동맥경화증이 감소됨이 보고되었으나[91], 골다공증 환자를 대상으로 한 대규모 연구(FREEDOM study)에서는 3년간의 denosumab 투여가 심혈관질환 합병증 발생 및 혈관 석회화 진행에 미치는 영향이 미미한 것으로 분석되었다[92]. 최근 만성 콩팥병 환자에서도 골다공증 위험요인이 있는 환자에서 골절 위험성을 평가하기 위하여 골밀도 검사를 시행할 것을 제안하였고[72], 신기능 감소에도 비교적 자유롭게 처방할 수 있는 denosumab의 개발로 인하여 많은 관련 임상의들의 관심과 기대가 많은 것은 사실이나, 국내 보험 인정기준으로 최대 투여 기간 제한이 있고, 장기간의 효과 및 합병증에 관한 연구 결과가 아직까지는 부족하고, 골절 및 사망 등의 주요한 임상 결과가 부족하므로, 아직까지 만성 콩팥병 환자에서 골다공증 진료 지침도 명확하게 합의되지 않은 상태이므로 혈관 석회화의 치료제로서의 역할은 보다 많은 연구가 필요할 것으로 생각된다.
결 론
지금까지 살펴 본 이전의 많은 생체외 및 생체내 연구에서 혈관평활근세포의 표현형 변화와 더불어 칼슘 침착이 혈관 석회화의 중요한 기전으로 생각되어지나, 현재까지는 이를 완화시키는 적절한 치료법이 개발되지 못한 상태이다. 만성 콩팥병 환자에서 중막 석회화가 병태생리적으로 중요하지만, 내막 석회화 및 심장판막 석회화 등과 동반되는 경우가 흔하고 이를 구분하여 각각의 치료 계획을 수립하는 것이 불가능할 것이므로, 다중 중재적 관리 전략이 필요하다고 생각된다. 즉, 혈관 석회화 유발요인을 피하기 위하여 칼슘계 인결합제, 과도한 활성 비타민 D 및 비타민 K 길항제 사용을 지양하고, 칼슘, 인, 비타민 D 및 PTH 등의 석회화 매개 변수의 균형을 잘 유지하도록 해야 하겠다. Sodium thiosulfate를 혈관 석회화의 치료제로서의 가능성을 타진한 소규모 선별 연구 결과가 흥미롭고, 최근 골다공증 치료제로 개발된 여러 약제가 만성 콩팥병 환자의 혈관 석회화에 미치는 영향에 관한 후속 연구가 이어진다면, 우리에게 보다 많은 정보를 제공할 수 있을 것으로 기대해 본다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






