


발작성 야간 혈색소뇨증과 골수형성이상종양을 동반한 환자에서 라불리주맙 중단 후 발생한 용혈 반동 및 반복적 혈전색전 사건: 증례 보고
- 박소연1, 이종미2, 김보현3, 김유진1, 박실비아1
Hemolytic Rebound and Recurrent Thromboembolic Event Following Ravulizumab Discontinuation in a Patient with PNH and MDS: A Case Report
- So Yeon Park1, Jong-Mi Lee2, Bohyun Kim3, Yoo-Jin Kim1, Silvia Park1
- Received March 30, 2025; Revised April 19, 2025; Accepted April 29, 2025;
- Abstract
-
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare clonal hematopoietic stem cell disorder characterized by complement-mediated hemolysis and thrombotic complications. The introduction of complement inhibitors has markedly improved survival outcomes by reducing intravascular hemolysis and thrombotic risk. We report the case of a 73-year-old man with PNH and myelodysplastic neoplasm (MDS) who developed severe thromboembolic and hemolytic events following the discontinuation of ravulizumab. His disease had previously evolved from aplastic anemia to MDS. Despite prior resolution of PNH-related thrombosis and maintained clinical stability, the cessation of treatment precipitated rebound hemolysis and multiple thrombotic events 8 months after discontinuation. These events included ischemic enterocolitis, necrotic gingivitis, ischemic epididymo-orchitis, and portal vein thrombosis. Resumption of complement inhibition with ravulizumab successfully halted the progression of PNH-related complications. This case emphasizes the life-threatening risks associated with the withdrawal of complement blockade in patients with a prior thromboembolic history and highlights the critical importance of continuous therapy, even in the context of bone marrow failure.
- INTRODUCTION
- INTRODUCTION
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare, acquired hematologic disorder characterized by chronic complement-mediated hemolysis, leading to anemia, profound fatigue, and thromboembolic events [1,2]. The prevalence is estimated at 10.4-38.1 cases per million individuals, with an incidence of 0.8-3.5 cases per million person-years, classifying PNH as an ultra-rare disease [3].PNH arises from an acquired PIGA gene mutation in hematopoietic stem cells, which disrupts glycosylphosphatidylinositol anchor synthesis [4]. This deficiency prevents key complement regulatory proteins, including CD55 and CD59, from attaching to red blood cells, rendering them susceptible to complement-mediated intravascular hemolysis (IVH) [5]. IVH is characterized by elevated reticulocyte counts, lactate dehydrogenase (LDH), and indirect bilirubin levels, along with decreased haptoglobin [6]. Importantly, elevated LDH is a strong predictor of thromboembolism, which accounts for 40-67% of PNH-related mortality, with a reported 10-year mortality rate of 24% in untreated patients [1,7-9].Complement inhibitors have revolutionized the treatment of PNH. Eculizumab, a C5 inhibitor, significantly improves clinical outcomes by preventing the formation of the membrane attack complex [10,11]. Ravulizumab, a next-generation C5 inhibitor with an extended half-life, provides prolonged C5 suppression and enhances survival outcomes [12,13]. Compared with untreated patients in the International PNH Registry, ravulizumab reduced mortality risk fivefold and increased the adjusted 4-year survival probability to 97.7%, reinforcing its long-term efficacy and safety [12].PNH may progress to bone marrow failure ( BMF), manifesting as aplastic anemia (AA) or myelodysplastic neoplasm (MDS) in 10-20% of cases [5,14-16]. Conversely, PNH clones can be present at the time of AA or MDS diagnosis [15,17]. Approximately two-third of patients with PNH clones are cytopenic and exhibit BMF, typically without clinical hemolysis [17]. However, these patients may experience evolving disease features and may develop clinically significant hemolysis at any point during the disease course [6]. When clinical manifestations of both BMF and PNH occur concurrently, management becomes particularly complex [2]. Although allogeneic hematopoietic stem cell transplantation (allo-HSCT) may be an option for younger and medically fit patients, it is often not feasible for elderly or medically unfit individuals, who may instead require supportive therapy for BMF and continued treatment with a complement inhibitor.In Korea, real-world clinical practice is often shaped by insurance policies, and coverage for ravulizumab excludes patients with acute myeloid leukemia or higher-risk MDS. Although ravulizumab may be appropriate for patients with lower-risk MDS, stringent regulations regarding highcost medications have created a psychological barrier to its prescription, occasionally resulting in drug discontinuation. We report the case of a 73-year-old patient with concomitant PNH and lower-risk MDS evolving from AA, who experienced catastrophic thromboembolic events following ravulizumab discontinuation. This case highlights the considerable risk associated with the withdrawal of complement inhibitor therapy, regardless of concomitant BMF.
- CASE REPORT
- CASE REPORT
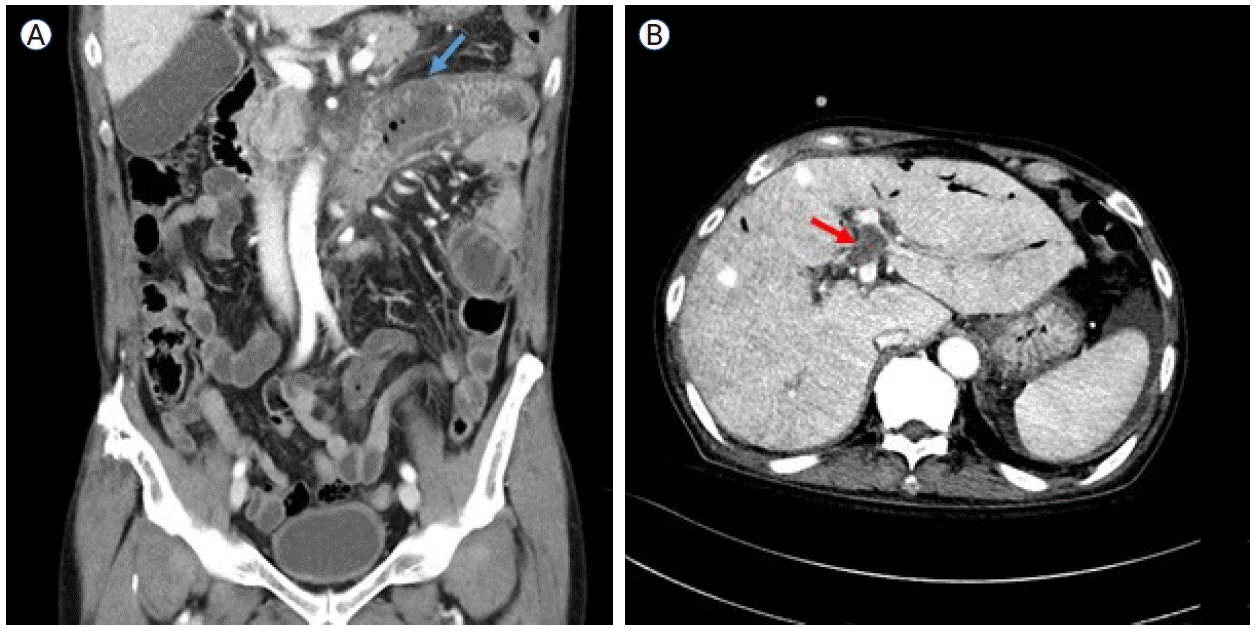
The patient was diagnosed with severe aplastic anemia (SAA) in 1989 and received antithymocyte globulin therapy, achieving a partial response. In October 2007, flow cytometry identified a PNH clone (75% in granulocytes and 51% in erythrocytes), and he was subsequently monitored for AA/PNH overlap syndrome. He remained clinically stable on oxymetholone, experiencing only two episodes of hemolytic crisis over 15 years, both of which resolved with methylprednisolone (mPD). This report was approved by the Institutional Review Board of The Catholic University of Korea (KC25ZASI0149). Informed consent for publication was obtained from the patient.In May 2022, the patient presented with fever and vomiting. Contrast-enhanced computed tomography (CT) and endoscopy revealed thrombosis in the middle hepatic vein and ischemic duodenitis, with the granulocyte PNH clone size increased to 91.63%. In July 2022, ravulizumab was initiated for PNH-related thrombosis in conjunction with rivaroxaban, with an LDH level of 1,594 U/L. By January 2023, the thrombus had resolved; however, worsening bicytopenia led to the introduction of cyclosporine for AA, which did not yield improvement. In June 2023, the patient presented with headache and gait disturbance, and a subdural hematoma was detected. Rivaroxaban was discontinued immediately, and burr hole trephination was performed. Despite well-controlled IVH on ravulizumab, progressive cytopenia prompted a bone marrow examination in December 2023. The examination (Fig. 1) revealed an MDS with low blasts with 50% cellularity, fewer than 1% blasts, and marked megakaryocytic dysplasia (50%). Molecular analysis demonstrated a normal karyotype and mutations in EP300 and TET2 gene. The patient was classified as having lower-risk MDS with a PNH clone (87.98% in granulocytes), with a Revised International Prognostic Scoring System (IPSS-R) score of 3.5 and a molecular IPSS score of -0.35, indicating moderately low risk. Due to uncertainty regarding insurance coverage in this setting, ravulizumab was discontinued after the final dose in December 2023. Thereafter, the patient remained stable with regular transfusions for 8 months following ravulizumab discontinuation. However, from May 2024 onward, LDH levels began to rise, reaching 1,000 U/L by August 2024.On August 19, 2024, the patient presented with abdominal pain and oral mucosal swelling. CT findings, together with dental examination results, suggested the development of ischemic enterocolitis (Fig. 2A) and necrotizing gingivitis, likely secondary to occult thrombosis. Progressive anemia (hemoglobin, 7.0 g/dL) was also noted, accompanied by gross hematuria and an elevated LDH level (787 U/L), consistent with IVH. Empirical antibiotics and intravenous steroid pulse therapy (mPD, 1 mg/kg) were initiated, resulting in symptom relief and stabilization of laboratory parameters. However, during steroid tapering, all symptoms and signs recurred, necessitating an increase in the steroid dosage. On September 19, the patient again reported abdominal pain with right upper quadrant tenderness and abdominal distension. A CT scan revealed a new non-malignant thrombus in the left portal vein (Fig. 2B) and the presence of ascites, which was further evaluated by a serum-ascites albumin gradient of 1.8 g/dL, indicating portal hypertension. On October 17, the patient developed epididymo-orchitis with a strong suspicion of ischemic infarction. Subsequently, prolonged steroid exposure led to the development of gastric and duodenal ulcers complicated by gastrointestinal bleeding, which required hemoclipping on three occasions.Due to recurrent, uncontrollable, and life-threatening thrombotic events, along with persistently elevated LDH levels (> 1.5 × baseline) associated with PNH (95.3% in granulocytes at that time), ravulizumab therapy was resumed following approval from the pre-review committee for PNH treatment. The induction dose was administered on October 28 (LDH, 491 IU/L), resulting in LDH normalization by November 4 (245 IU/L). However, an episode of Pseudomonas aeruginosa pneumonia delayed the maintenance dose, which was eventually administered on December 2. Since then, LDH levels have remained within the normal range, and follow-up imaging has demonstrated a reduction in the size of the portal vein thrombus, despite the absence of anticoagulation therapy due to thrombocytopenia. Furthermore, ischemic enteritis has improved, as evidenced by decreased duodenal wall thickening. The patient continues on ravulizumab maintenance therapy with regular outpatient follow-up.
- DISCUSSION
- DISCUSSION
PNH is a chronic, complement-mediated hemolytic disorder associated with a substantial risk of thrombotic complications [1,5]. Historically, thromboembolic events have been the leading cause of mortality in untreated PNH, and the introduction of complement inhibitors, including eculizumab and ravulizumab, has markedly reduced this risk [12]. Active hemolysis, reflected by elevated LDH levels, is a key risk factor for the development of thromboembolic events. Moreover, even in the absence of overt hemolytic manifestations, the presence of PNH clones has been associated with an increased thrombotic risk in patients with MDS compared with those without PNH clones [15]. This case illustrates the severe consequences of discontinuing complement inhibition in a patient with PNH-MDS overlap syndrome, despite the prior resolution of PNH-related thromboembolic events and clinical stability under ravulizumab therapy.The pathophysiology of PNH-associated thrombosis is multifactorial, involving IVH, free hemoglobin-mediated nitric oxide depletion, endothelial dysfunction, platelet activation, and complement-driven inflammation [1,5,18]. These mechanisms collectively create a markedly prothrombotic state. Given the high thrombotic risk in PNH, sustained complement inhibition remains the cornerstone of management to prevent thromboembolic complications. Discontinuation of C5 inhibitor therapy is uncommon; however, when it occurs, it may precipitate rebound hemolysis [19] and rapid clinical deterioration. In this case, withdrawal of ravulizumab led to marked elevations in hemolysis markers and severe thrombo-ischemic events, including ischemic enterocolitis, necrotizing gingivitis, ischemic epididymo-orchitis, and portal vein thrombosis, likely secondary to vascular compromise. Notably, clinical deterioration manifested approximately 8 months after discontinuation of ravulizumab, well beyond its elimination half-life of approximately 8 weeks.Beyond the thromboembolic risk of PNH, this case highlights the strong clinical association between PNH and BMF and demonstrates how these conditions may evolve throughout the disease course. In this patient, the initial manifestation was pancytopenia, consistent with SAA criteria. Following immunosuppressive therapy, cytopenia improved; however, such treatment may create a selective environment that permits PNH clone expansion over time [20], leading to IVH and subsequent PNH-related thromboembolic events. With ravulizumab therapy, IVH stabilized, and the thrombus resolved; however, cytopenia worsened, and lower-risk MDS was newly diagnosed.At this stage, physicians may encounter multiple clinical challenges, and therapeutic decisions are not as straightforward as in cases of classic PNH or isolated BMF with a minor PNH clone lacking hemolytic activity. Both thrombotic risk from PNH and hemorrhagic risk due to MDS-related thrombocytopenia can coexist. Considering that the patient is 73 years old and has a history of gastrointestinal bleeding with a concurrent portal vein thrombus, curative treatment with allo-HSCT is not feasible. For anemia management, erythropoiesis-stimulating agents may be considered in lower-risk MDS patients with serum erythropoietin levels below 500 mIU/mL. However, this approach carries a potential increase in thrombotic risk. Overall, the management of PNH-MDS overlap syndrome is particularly challenging, especially when both disease phenotypes are clinically active, requiring individualized and carefully balanced therapeutic strategies.In conclusion, this case emphasizes the severe and potentially catastrophic consequences of discontinuing complement inhibition, even after the apparent resolution and stabilization of PNH-related complications. This remains true regardless of concomitant BMF, where cytopenia may be profound.
- Conflicts of Interest
- Conflicts of Interest
-
CONFLICTS OF INTEREST No potential conflict of interest relevant to this article was reported.
- Notes
- Notes
-
FUNDING None.
- Notes
- Notes
-
AUTHOR CONTRIBUTIONS Conceptualization: Park S.
Data curation: Park SY, Park S.
Visualization: Lee JM, Kim B.
Writing - original draft: Park SY.
Writing - review & editing: Park SY, Kim YJ, and Park S.
- Notes
- Notes
-
ACKNOWLEDGEMENTS None.
Figure 1.
Representative bone marrow aspirate smears stained with Wright-Giemsa demonstrate dyserythropoiesis (red arrows) and dysmegakaryopoiesis (blue arrow) at × 200 magnification. Dyserythropoiesis is characterized by multinuclearity and karyorrhexis of erythroid precursors, whereas dysmegakaryopoiesis shows abnormal nuclear segmentation.

Figure 2.
Computed tomography images of the patient’s abdomen. (A) Short segmental wall thickening in the proximal jejunum and the third and fourth portions of the duodenum (red arrow), suggestive of bowel ischemia secondary to occult thrombosis. (B) A newly developed thrombus in the umbilical segment of the left portal vein (blue arrow), accompanied by ascites.
- References
- References
REFERENCES
1. Hill A, Kelly RJ, Hillmen P. Thrombosis in paroxysmal nocturnal hemoglobinuria. Blood 2013;121:4985–4996.
[Article] [PubMed]2. Kelly RJ, Holt M, Vidler J, et al. Treatment outcomes of complement protein C5 inhibition in 509 UK patients with paroxysmal nocturnal hemoglobinuria. Blood 2024;143:1157–1166.
[Article] [PubMed]3. Richards SJ, Painter D, Dickinson AJ, et al. The incidence and prevalence of patients with paroxysmal nocturnal haemoglobinuria and aplastic anaemia PNH syndrome: a retrospective analysis of the UK's population-based haematological malignancy research network 2004-2018. Eur J Haematol 2021;107:211–218.
[Article] [PubMed]4. Mahoney JF, Urakaze M, Hall S, et al. Defective glycosylphosphatidylinositol anchor synthesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood 1992;79:1400–1403.
[Article] [PubMed]5. Hillmen P, Lewis SM, Bessler M, Luzzatto L, Dacie JV. Natural history of paroxysmal nocturnal hemoglobinuria. N Engl J Med 1995;333:1253–1258.
[Article] [PubMed]6. Risitano AM, de Latour RP. How we('ll) treat paroxysmal nocturnal haemoglobinuria: diving into the future. Br J Haematol 2022;196:288–303.
[Article] [PubMed]7. Jang JH, Kim JS, Lim CTK, et al. Impact of lactate dehydrogenase and hemoglobin levels on clinical outcomes in patients with paroxysmal nocturnal hemoglobinuria: results from the National Korean PNH Registry. J Korean Med Sci 2024;39:e81.
[Article] [PubMed] [PMC]8. Jang JH, Kim JS, Yoon SS, et al. Predictive factors of mortality in population of patients with paroxysmal nocturnal hemoglobinuria (PNH): results from a Korean PNH Registry. J Korean Med Sci 2016;31:214–221.
[Article] [PubMed] [PMC]9. Griffin M, Hillmen P, Munir T, et al. Significant hemolysis is not required for thrombosis in paroxysmal nocturnal hemoglobinuria. Haematologica 2019;104:e94–e96.
[Article] [PubMed]10. Brodsky RA, Young NS, Antonioli E, et al. Multicenter phase 3 study of the complement inhibitor eculizumab for the treatment of patients with paroxysmal nocturnal hemoglobinuria. Blood 2008;111:1840–1847.
[Article] [PubMed]11. Kim JS, Lee JW, Kim BK, Lee JH, Chung J. The use of the complement inhibitor eculizumab (Soliris®) for treating Korean patients with paroxysmal nocturnal hemoglobinuria. Korean J Hematol 2010;45:269–274.
[Article] [PubMed] [PMC]12. Kulasekararaj A, Brodsky R, Schrezenmeier H, et al. Ravulizumab demonstrates long-term efficacy, safety and favorable patient survival in patients with paroxysmal nocturnal hemoglobinuria. Ann Hematol 2025;104:81–94.
[Article] [PubMed] [PMC]13. Lee JW, de Fontbrune FS, Lee LWL, et al. Ravulizumab (ALXN1210) vs eculizumab in adult patients with PNH naive to complement inhibitors: the 301 study. Blood 2019;133:530–539.
[Article] [PubMed]14. Morado M, Sandes AF, Colado E, et al. Diagnostic screening of paroxysmal nocturnal hemoglobinuria: prospective multicentric evaluation of the current medical indications. Cytometry B Clin Cytom 2017;92:361–370.
[Article] [PubMed]15. Fattizzo B, Ireland R, Dunlop A, et al. Clinical and prognostic significance of small paroxysmal nocturnal hemoglobinuria clones in myelodysplastic syndrome and aplastic anemia. Leukemia 2021;35:3223–3231.
[Article] [PubMed] [PMC]16. Galili N, Ravandi F, Palermo G, et al. Prevalence of paroxysmal nocturnal hemoglobinuria (PNH) cells in patients with myelodysplastic syndromes (MDS), aplastic anemia (AA), or other bone marrow failure (BMF) syndromes: interim results from the EXPLORE trial. J Clin Oncol 2009;27 Suppl 15:7082.
[Article]17. Babushok DV. When does a PNH clone have clinical significance? Hematology Am Soc Hematol Educ Program 2021;2021:143–152.
[Article] [PubMed] [PMC]18. Brodsky RA. Paroxysmal nocturnal hemoglobinuria. Blood 2014;124:2804–2811.
[Article] [PubMed] [PMC]