


2025 유럽호흡기학회/미국흉부학회 간질성 폐렴 분류 개정안: 주요 변경 사항
- 김경훈1, 송진우2
2025 ERS/ATS Update of the Classification of Interstitial Pneumonias: Key Changes
- Kyung Hoon Kim1, Jin Woo Song2
- Received December 12, 2025; Revised January 17, 2026; Accepted January 24, 2026;
- Abstract
-
Interstitial pneumonia is a heterogeneous group of diffuse parenchymal lung diseases characterized by variable combinations of inflammation and fibrosis involving the interstitium, airways, and alveolar spaces. The classification of interstitial pneumonia has undergone a major update for the first time in 12 years since 2013. The 2025 European Respiratory Society/American Thoracic Society (ERS/ ATS) update introduces an integrated framework that extends beyond idiopathic entities to include secondary causes such as connective tissue disease and hypersensitivity pneumonitis. This update classifies interstitial pneumonias into ‘interstitial patterns’ and ‘alveolar filling patterns’ based on the predominant site of involvement, with interstitial patterns further subclassified into ‘fibrotic’ and ‘nonfibrotic’. Furthermore, it newly defines and reintroduces the ‘bronchiolocentric interstitial pneumonia’ pattern to replace the traditional ‘hypersensitivity pneumonitis pattern’ and renames the terms ‘acute interstitial pneumonia’ and ‘desquamative interstitial pneumonia’ to ‘diffuse alveolar damage’ and ‘alveolar macrophage pneumonia’, respectively, to more accurately reflect the underlying pathophysiology. Importantly, the update distinguishes morphologic patterns from multidisciplinary discussion diagnoses and formalizes the concept of diagnostic confidence, presenting a system of ‘confident’, ‘provisional’, or ‘unclassifiable interstitial lung disease’. Although this update is based on expert consensus, the new classification framework can provide a more comprehensive and practical approach for clinicians managing patients with complex interstitial pneumonias. This review summarizes these key changes and their clinical implications.
- 서 론
- 서 론
간질성 폐렴(interstitial pneumonia, IP)은 폐의 간질과 기도, 폐포를 침범하여 염증과 섬유화를 유발하는 약 200여 종의 다양한 질환을 아우르는 포괄적인 용어이다[1]. 간질성 폐렴은 임상 양상과 영상, 병리 소견이 다양하고 자가면역 질환, 환경성, 흡입 노출, 약제, 유전적 요인 등 다양한 이차적인 원인에 의해서도 발생할 수 있다. 때로는 원인을 찾지 못하는 경우도 있으며 동일한 환자에서도 시간 경과에 따라 질병의 양상이 달라져 진단과 치료가 어려운 질환군이다.2002년 미국흉부학회(American Thoracic Society, ATS)/유럽호흡기학회(European Respiratory Society, ERS)가 특발성 간질성 폐렴 분류에 대한 국제 다학제 합의문을 발표하며 처음으로 간질성 폐렴을 특발성 폐섬유증(idiopathic pulmonary fibrosis, IPF), 비특이 간질성 폐렴(non-specific interstitial pneumonia, NSIP), 특발성 기질화 폐렴(cryptogenic organizing pneumonia, COP) 등 7개로 분류하는 시도를 하였다[2]. 이후 2013년 개정에서는 2002년에는 잠정적 진단으로 남겨 두었던 NSIP를 독립적인 질환으로 인정하였고 흉막폐실질 섬유탄력증(pleuroparenchymal fibroelastosis, PPFE)을 추가하였으며 혼합 패턴과 질병 행동의 개념을 도입하고 다학제적 논의(multidisciplinary discussion, MDD)를 진단의 필수 요소로 제시하였다[3]. 그러나 기존 분류 체계는 특발성 질환에만 국한되어 있어 결체조직 질환 관련 간질성 폐질환(interstitial lung disease, ILD), 과민성 폐렴(hypersensitivity pneumonitis, HP), 약제 유발 ILD 등의 이차성 원인에 의한 간질성 폐렴은 포함하지 못한다는 한계가 있었다. 본고에서는 최근 12년 만에 새로이 발표된 2025 ERS/ATS 간질성 폐렴 분류 개정안의 주요 내용을 소개하고 임상적 함의를 고찰하고자 한다.
- 본 론
- 본 론
- 새로운 분류 체계와 4가지 주요 변화
- 새로운 분류 체계와 4가지 주요 변화
간질성 폐렴 전체로 분류 체계 확장
간질성 폐렴 전체로 분류 체계 확장
첫 번째 변화이자 가장 큰 변화는 특발성과 이차성 간질성 폐렴의 통합이다. 특발성에 국한되어 있던 기존 분류 체계에서 간질성 폐렴 전체를 분류하는 체계를 이번 개정안에서 제시하였다. 이는 질병의 초기에는 원인이 불분명한 경우가 많고 호흡세기관지염 연관 간질성 폐질환(respiratory bronchiolitis-interstitial lung disease, RB-ILD)과 같은 일부 질환은 특발성이라 간주되기 어려웠으며 특발성이라 불리는 질환조차 기저 원인이 있을 가능성이 있기 때문이다[4]. 이러한 접근은 원인이 불분명한 경우에도 형태학적 패턴에 기반한 잠정적 분류를 가능하게 하여 향후 임상 경과에 따라 진단을 수정하거나 구체화하는 것이 용이하다.새로운 대분류 체계 도입: 간질성 질환과 폐포 충만 질환
새로운 대분류 체계 도입: 간질성 질환과 폐포 충만 질환
두 번째, 간질성 폐렴 전체를 간질성(interstitial) 패턴과 폐포 충만(alveolar filling) 패턴의 두 축으로 대분류하였다. 간질성 패턴은 다시 섬유화 유무에 따라 섬유성(fibrotic)과 비섬유성(non-fibrotic)으로 분류하였다. 형태학적 분류의 세분화와 함께 질병 진행 양상(anticipated disease behaviour)의 중요성도 강조되었는데, 이는 동일 패턴을 보이는 간질성 폐렴 내에서도 질병의 진행과 경과는 다를 수 있음을 시사한다. 이러한 관점은 최근 발표된 임상 진료지침에서도 진행성 폐섬유증(progressive pulmonary fibrosis)으로 제안되어 다루어졌다[5].새로운 하위 범주 및 용어 도입
새로운 하위 범주 및 용어 도입
세 번째, 오해의 소지가 있거나 병리학적 기전과 일치하지 않는 용어들을 수정하고 새로운 개념을 도입하였다. 먼저 세기관지 중심 간질성 폐렴(bronchiolocentric interstitial pneumonia, BIP)이라는 용어를 공식적으로 도입하였다. 병리 및 영상 소견에서 세기관지 중심 패턴을 보이는 환자의 상당수가 HP가 아닌 진단이었다. 즉, 흡인, 결체조직 질환, 약제 관련 간질성 폐렴 등 다른 간질성 폐렴에서도 기도 중심의 병리학적/영상학적 패턴이 관찰될 수 있다[6,7]. 따라서 HP 패턴이라는 용어 대신 병변의 해부학적 위치를 객관적으로 기술하는 BIP 패턴을 usual interstitial pneumonia (UIP), NSIP와 함께 3대 주요 패턴으로 재정의하였다. 만약 철저한 원인 조사 후에도 원인을 찾지 못한 경우 특발성 BIP라는 잠정 진단을 부여하고 추적 관찰하게 된다. 또한 부정확한 용어였던 급성 간질성 폐렴(acute interstitial pneumonia, AIP)을 특발성 미만성 폐포 손상(diffuse alveolar damage, DAD)으로 변경하였고 박리성 간질성 폐렴(desquamative interstitial pneumonia, DIP)도 폐포 대식세포 폐렴(alveolar macrophage pneumonia, AMP)으로 변경하였다.진단 신뢰도 개념의 도입
진단 신뢰도 개념의 도입
네 번째, 진단 신뢰도와 잠정 진단을 공식화하였다. 실제 임상 현장에서는 고령, 동반 질환, 환자의 거부 등 다양한 이유로 폐 생검을 하지 못하는 경우가 흔하다. 또한 확정적 진단을 제시할 수 있는 단일 검사가 있지 않기 때문에 진단의 확률을 명시하는 것이 중요하다. 이에 MDD를 거친 후 특정 진단의 신뢰도가 90% 이상인 경우는 확정 진단(confident diagnosis), 50-89% 수준인 경우에는 잠정 진단(provisional diagnosis)의 용어를 쓰는 것을 공식적으로 제시하였다. 예를 들어 전형적인 HP의 영상 소견을 보이지만 명확한 항원을 찾지 못한 경우 잠정적 HP 또는 특발성 BIP로 잠정 진단하고 치료를 시작할 수 있다. 또한 신뢰도가 50% 이하일 때는 분류 불가능(unclassifiable) ILD로 분류할 것을 제시하였다.
개정된 분류 접근법에서 가장 중요하면서도 기본이 되는 점은 영상/병리 패턴을 기반으로 환자를 분류하고 그 다음 MDD를 통해 환자를 진단한다는 점이다(Fig. 1; Table 1). 즉 패턴과 다학제 진단을 구분하였다. 예를 들어 NSIP는 패턴이고 특발성 NSIP는 다학제 진단이다. 이번 개정에서는 네 가지 주요 변화가 있었다.- 진단 접근
- 진단 접근
대부분의 간질성 폐렴은 특이적인 진단 검사가 없으며 여러 영역(임상, 영상, 병리 등)의 정보를 통합하여 진단이 이루어진다. 진단의 시작은 흉부 전산화단층촬영(computed tomography, CT)에서 보이는 영상 패턴을 확인하는 것에서부터 시작한다. 먼저 간질성 질환과 폐포 충만 질환을 구별하는 것으로 시작하며 이후 개별 패턴으로 세분화한다(Fig. 1; Table 2). 이후 영상 소견과 임상 정보, 혈액 검사 소견을 통합하여 진단하게 되는데 이것만으로 확정 진단(confident diagnosis)을 내리고 치료 결정을 할 수 있다. 다만 임상, 영상 및 혈액 검사 소견 등을 종합한 후에도 진단이나 치료 방침이 불확실할 경우 수술적 폐 생검과 같은 침습적인 조직 검사를 고려할 수 있다. 또한 진단 과정 전반에 있어 진단 신뢰도(diagnostic confidence)를 기록할 것을 권고하고 있다.- 주요 병리학적 및 영상학적 패턴
- 주요 병리학적 및 영상학적 패턴
간질성 패턴
간질성 패턴
간질성 패턴은 UIP, NSIP, BIP, DAD, PPFE, lymphoid interstitial pneumonia (LIP)로 분류하였다(Table 1). 이 중 가장 흔한 패턴은 UIP, NSIP, BIP이다. 이들 패턴은 대개 진행성 폐섬유증과 불량한 예후와 연관되어 있는 경우가 많다. NSIP와 BIP는 섬유화가 적고 세포성이 더 많은 형태로도 나타나고, 이 경우 예후는 더 양호할 수 있다. 이번 개정에서 중요한 변화인 BIP와 DAD에 대해서 자세히 알아본다.세기관지 중심 간질성 폐렴
세기관지 중심 간질성 폐렴
BIP라는 용어는 이전부터 있던 용어[8]이나 이번 개정에는 새로운 정의를 적용하여 다시 도입할 것을 제안하였다. 즉, BIP를 주로 기도 중심으로 분포하는 간질성 폐렴을 나타내는 병리학적, 영상학적 패턴을 정의하는 상위 개념으로 사용하도록 하였다. 이러한 패턴은 HP에서만 보이는 것이 아니고 connective tissue disease (CTD)-ILD, 흡인, 흡입 노출, 약제 노출 등 매우 다양한 상황에서 관찰될 수 있다[6,9]. 따라서 HP라는 용어는 영상 또는 병리적 패턴이 아닌 MDD를 통해 확정된 임상 진단에 한해서만 사용할 것을 제안하였다. 영상이나 병리에서 HP pattern이라고 기술할 경우 마치 이미 MDD에서 HP가 확정된 것처럼 오해될 수 있으며, 이는 정확하지 않은 임상 진단을 야기할 수 있기 때문이다.BIP는 간질의 염증과 섬유화가 혼재되어 있을 수 있다(Fig. 2A-C). 비섬유성(또는 세포성) BIP의 병리학적 특징은 세기관지를 둘러싸는 림프구-우세 만성 염증 소견이며 비괴사성 육아종이 동반될 수 있다. 섬유성 BIP는 세기관지 중심의 섬유화를 특징으로 하며 인접한 폐포에서는 세기관지 주위 화생(peribronchiolar metaplasia)이 종종 관찰된다. 비섬유성 BIP의 전형적인 CT 소견은 세기관지 주위 염증을 시사하는 중심소엽성 결절과 소기도 폐쇄를 시사하는 모자이크 음영 및 공기 저류이다(Fig. 2D, E). 섬유성 BIP에서는 간유리음영, 망상 음영, 견인기관지 확장, 삼상 폐밀도 양상, 호기시 공기 저류의 소견이 기관지혈관 주위 분포를 보인다(Fig. 2F) [9]. 이러한 소견들은 임상적 상황이 맞다면 HP를 강력하게 시사하는 소견들이지만 CTD-ILD 등 다른 원인에 의한 간질성 폐렴에서도 관찰될 수 있다[10].BIP 도입의 목적은 HP를 포함한 다양한 기도 중심 간질성 폐렴을 보다 정확하게 구분하고 HP로 단정할 수 없는 환자를 체계적으로 분류하기 위한 것이다. 원인이 분명하지 않은데 BIP 패턴을 보이는 경우 특발성 BIP라는 잠정 진단(provisional diagnosis)을 사용할 것을 제안하였다. 최종 진단으로서의 HP와 잠정 진단으로서의 특발성 BIP를 구분하는 데에는 항원 노출 가능성에 대한 평가가 필요하다. 또한 일부 환자에서는 원인 항원이 확인되지 않더라도 고전적인 조직학적 영상학적 소견을 바탕으로 HP로 확신할 수 있다.하지만 BIP 패턴을 도입하는 데 반대가 없었던 것은 아니다. 위원회의 80% 이상은 BIP를 주요 패턴으로 포함하고 특발성 BIP를 잠정 진단으로 도입하는 데 찬성하였으나 네 명의 위원이 개정안의 공동 저자에서 제외되기를 요청하였다[1].미만성 폐포 손상
미만성 폐포 손상
미만성 폐포 손상(DAD)은 급성 폐 손상의 대표적인 병리 패턴이다. 초기의 급성기는 폐포 중격 손상이 주로 일어나며 후기의 조직화기(organizing phase)에서는 폐포 충만이 나타나는 특징을 보인다(Fig. 3A, B). 급성기에는 간질 부종, 폐포 증식, 유리질막 형성으로 폐포 격벽의 비후가 일어난다. 이후 조직화기로 진행하면서 느슨한 섬유조직이 폐포벽에 자리하게 되는데, 이는 폐포 내부에 결체조직이 형성되는 기질화 폐렴(organizing pneumonia, OP)과 구분되는 점이다[3]. CT에서는 양측성 간유리음영(국소적으로 정상 부위를 동반), 폐경화 소견(중력 의존), 견인성 기관지 또는 세기관지 확장증이 특징적으로 관찰된다(Fig. 3C) [11,12]. 임상적으로는 급성 또는 아급성 호흡 부전으로 이어지며 높은 사망률을 보인다. DAD는 급성 호흡곤란증후군(acute respiratory distress syndrome)의 가장 주요한 조직학적 소견으로 감염, 약제, 흡입 손상, 결체조직 질환(특히 염증성 근육병증) 등 매우 다양한 원인에 의하여 발생한다. 특발성 간질성 폐렴의 급성 악화에서도 가장 흔한 병리 소견이다[13]. 특발성 DAD는 이전에 AIP로 불렸으나[3] 다른 간질성 폐렴들도 급성으로 나타날 수 있기 때문에 이번 개정에서 DAD로 변경되었다.폐포 충만 패턴
폐포 충만 패턴
폐포 대식세포 폐렴
폐포 대식세포 폐렴
AMP는 폐포 내에 대식세포가 광범위하게 축적되는 현상을 기술하는 용어로 기존의 DIP를 대체한다[14]. AMP의 대부분은 흡연과 관련이 있지만 이차성 원인과 연관되거나 특발성일 수도 있다[14,15]. AMP는 폐포 공간 내에서는 균일한 분포를 보이는 미만성 폐 침범의 양상을 보이며, 폐포벽은 섬유화되거나 다양한 염증 소견을 보이기도 한다(Fig. 4A). 많은 환자들에서 RB-ILD와 AMP의 특징이 모두 보이는 경우가 흔한데, 우세한 패턴에 따라 명명하게 된다. UIP나 NSIP와 혼재되기도 한다. 영상에서는 광범위한 간유리음영이 전형적인 소견이며 RB-ILD보다 더 광범위하고 융합된 형태를 보인다(Fig. 4B, C). 기타 CT 소견으로는 섬유화, 낭종, 폐기종이 포함될 수 있으나 벌집 모양은 드물다[15].복합 패턴
복합 패턴
- 분류 불가능 간질성 폐질환
- 분류 불가능 간질성 폐질환
철저한 다학제적 평가와 진단에도 불구하고 일부 환자들에서는 여전히 분류가 불가능한 경우가 있다. 이러한 분류 불가능 ILD의 전체 유병률은 11.9%, MDD 후에도 9.5%로 보고된 바 있다[19]. 이는 폐 생검을 하지 못한 경우처럼 임상 정보가 부족한 경우, 영상은 UIP 소견이나 병리는 NSIP 소견이 나오는 경우처럼 정보의 불일치를 보이는 경우, 정말로 분류가 어려운 경우(truly unclassifiable)들이 포함된다[20]. 한 국제 전문가 그룹에서는 폐 생검 여부와 상관없이 충분한 MDD 이후에도 50% 이상의 확신(confidence)을 가지지 못한 경우 분류 불가능 간질성 폐렴으로 정의할 것을 제안하였으며 진단의 신뢰도가 90% 이상인 경우는 확정 진단(confident diagnosis), 51-89%면 잠정 진단(provisional diagnosis)으로 간주하였다[21].- 간질성 폐렴에서의 분자생물학적 진전
- 간질성 폐렴에서의 분자생물학적 진전
이번 개정안에서는 영상학적, 병리학적 패턴에 기반한 분류를 유지하면서도 분자생물학적인 발전이 향후 진단 및 분류 체계에 미칠 영향에 대해서도 강조하였다. 가장 잘 알려진 유전적 소견인 단일 염기 다형성은 MUC5B 프로모터 변이(rs357805950)이나 아시아인에게는 드물어서 실제 임상 적용에는 한계가 있다[22,23]. IPF 환자와 정상 대조군의 폐조직의 전사체 분석을 통해 UIP 패턴을 식별할 수 있었으며[24] 말초 혈액 백혈구의 텔로미어 길이가 ILD 환자의 생존과 폐활량 감소와 연관된다는 연구들도 축적되고 있다[24]. 다양한 혈청 바이오마커들이 IPF를 정상인과 구별하거나 예후를 예측하는 데 유용하다는 연구들이 있으나[25-27] 실제 임상적 유용성은 아직 명확하지 않다. 이번 개정안은 이러한 분자 진단 기술들이 현재의 분류 체계에는 포함되지 않지만 향후 진단 신뢰도를 향상시키고 질병의 이질성을 해석하는 데 중요한 역할을 할 것으로 전망하였다.- 제한점
- 제한점
이번 개정안에서는 전체 간질성 폐렴으로 분류 체계를 확장하고 새로운 용어를 도입하는 등의 많은 변화가 있다. 하지만 이러한 개정 전체가 근거에 기반하지 않고 전문가 합의에 의한 개정이라는 점이 가장 큰 제한점이다. 또한 BIP를 독립된 주요 패턴으로 도입하였으나 병리학적, 영상학적 정의가 확실하지 않고, 비교적 최근인 2020년에 발표된 과민성 폐렴에 대한 국제 임상 진료지침의 정의 및 용어와 많은 부분들이 상충되어 호흡기내과, 영상의학과, 병리과 의사들 사이에 혼란을 가중시킬 수 있다. 이와 더불어 BIP에 대한 위원들 간의 불일치로 4명의 위원이 공동 저자에서 제외되었다는 사실 자체도 제한점이 되겠다. 마지막으로 진단 신뢰도를 도입할 때 90%, 50% 등 신뢰도의 평가는 어떻게 할지 모호하여 진단에 혼란을 가중시킬 가능성도 없지 않다.
개정안의 세부 내용을 살펴보기 전에 이해를 돕기 위하여 몇 가지 전제 사항을 설명하고자 한다. 첫째, ‘간질성 폐렴’이라는 용어를 유지하였다. 이는 2002년과 2013년 분류와의 연속성을 유지하면서 간질성 질환뿐만 아니라 폐포 충만(alveolar filling) 질환까지 포함할 수 있기에 일부 제한점에도 불구하고 유지하기로 하였다. 또한 ‘간질성 폐질환’도 쓰도록 하였다. 둘째, 이번 개정안에서는 질병의 분류에 초점을 맞추었으며 치료를 다루지는 않았다. 셋째, 이번 개정안은 근거 중심 진료지침이 아닌 전문가 합의문(consensus statement)이다. 32명의 전문가가 참여하여 합의를 도출하였으며 70% 이상의 동의가 있을 때 합의된 것으로 간주하였다.
- 결 론
- 결 론
이번 개정안은 2013년 이후 12년 만의 개정으로 가장 큰 변화는 특발성 간질성 폐렴뿐만 아닌 전체 간질성 폐렴을 분류하려는 틀을 제시하였다는 점이다. 또한 BIP나 AMP 같은 새로운 용어를 제안하였고 전체 간질성 폐렴을 간질성 패턴과 폐포 충만 패턴으로 하위 분류할 것을 제시하였다. 그러나 이번 개정안은 전문가 합의에 기초를 두고 있어 BIP와 같은 새로운 정의의 근거가 충분치 않으며 국제 HP 진료지침과 혼동되어 실제 임상 현장에서는 혼란이 지속될 가능성이 없지 않다. 이러한 점에서 이번 개정안은 의미 있는 진전으로 생각되지만 여전히 과도기적이라 생각된다. 종합하면 이번 개정안은 전체 간질성 폐렴을 분류하는 시도로 이전 분류 체계에서 큰 변화가 있다. 향후 연구를 통해 이번 개정안이 제안한 분류 체계의 유용성과 정확성이 입증되어야 할 것으로 생각된다.
- Conflicts of Interest
- Conflicts of Interest
-
CONFLICTS OF INTEREST No potential conflict of interest relevant to this article was reported.
- Notes
- Notes
-
FUNDING None.
- Notes
- Notes
-
AUTHOR CONTRIBUTIONS Kyung Hoon Kim contributed to the conceptualization, literature search, drafting, and revision of the manuscript.
Jin Woo Song supervised the study, provided conceptual guidance, critically revised the manuscript for important intellectual content, and approved the final version of the manuscript.
- Notes
- Notes
-
ACKNOWLEDGEMENTS None.
Figure 1.
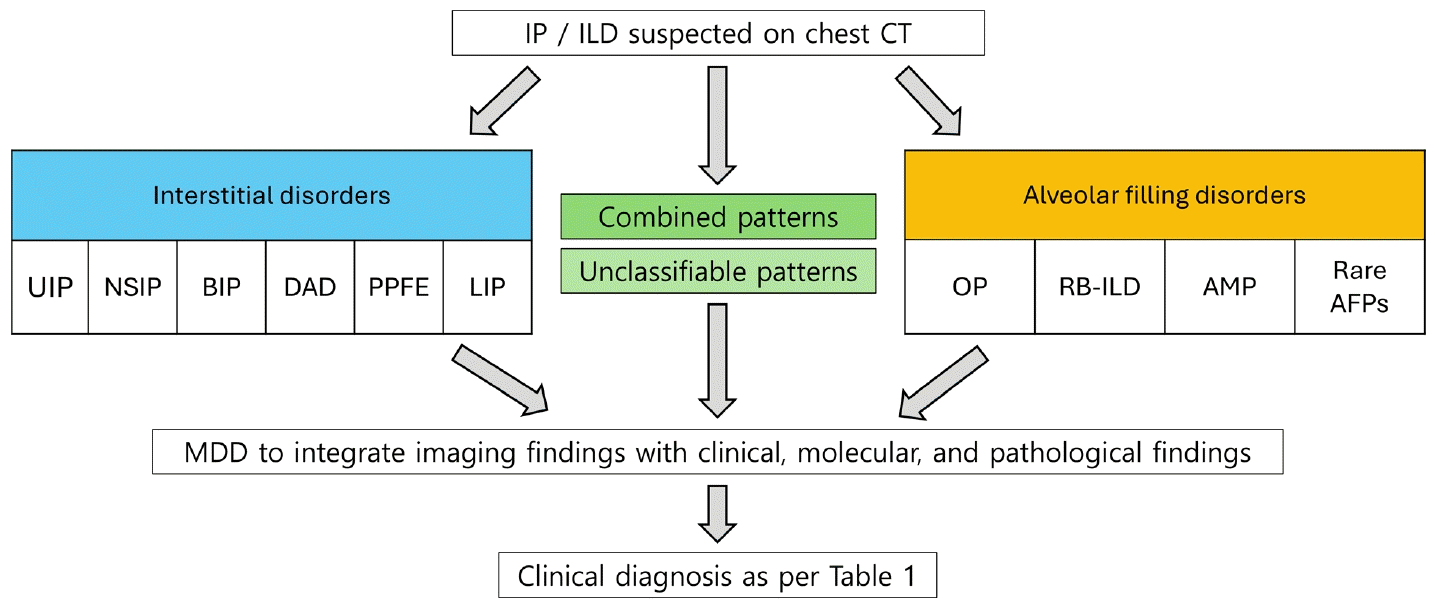
Figure 2.
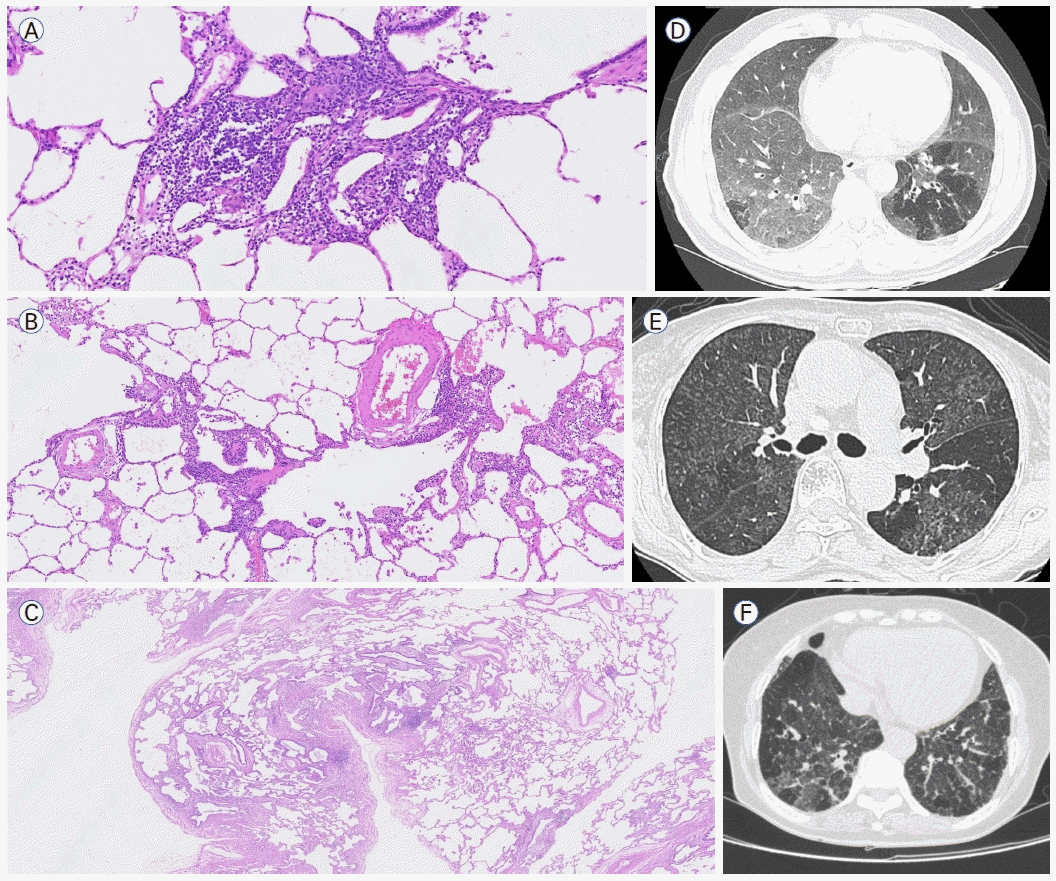
Figure 3.

Figure 4.
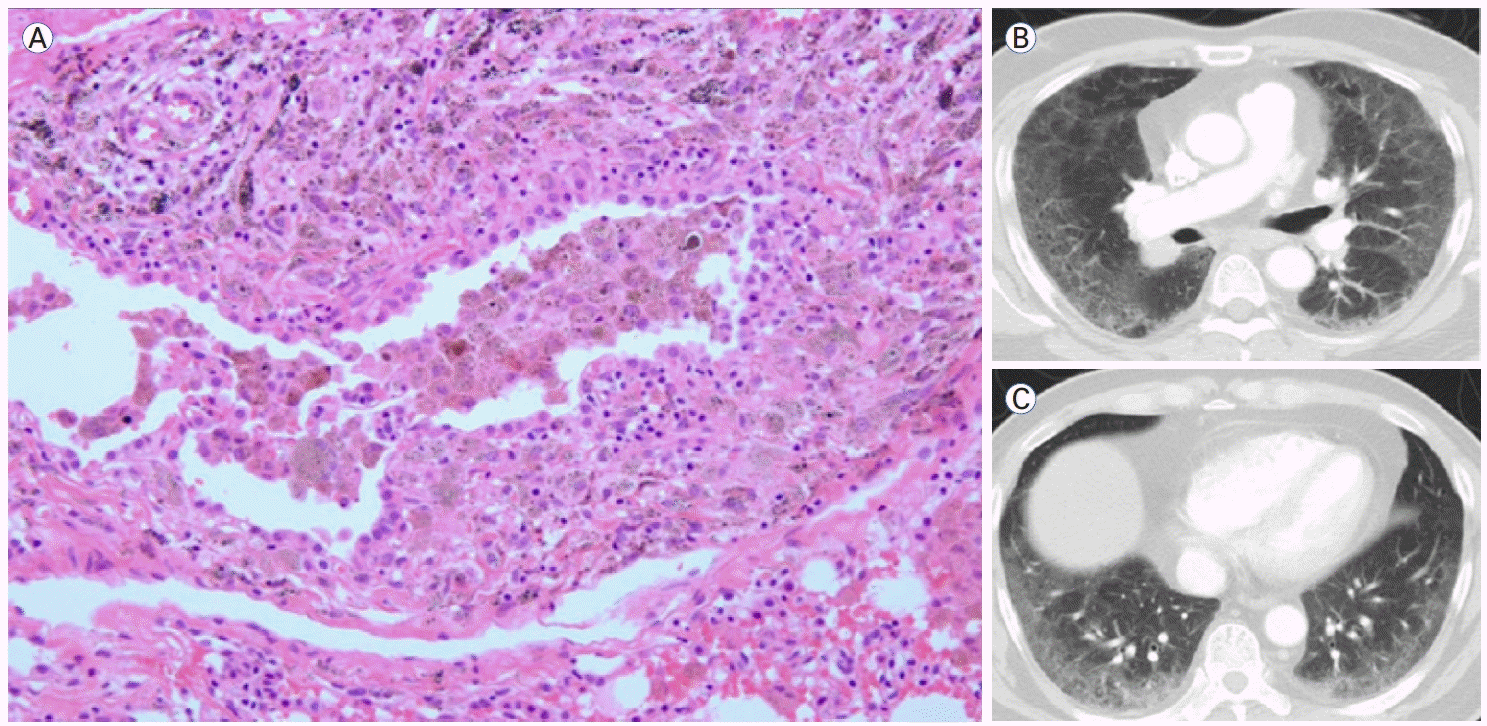
Table 1.
| Morphologic patterns by pathology and imaging |
Major clinical-radiologic-pathologic diagnoses |
|
|---|---|---|
| Secondarya | Primary/idiopathic | |
| Interstitial patterns | ||
| Usual interstitial pneumonia (UIP) | Secondary UIP (e.g., CTD, HP, medications) | Idiopathic pulmonary fibrosis (Idiopathic UIP) |
| Nonspecific interstitial pneumonia (NSIP) | Secondary NSIP (e.g., CTD >>> HP, medications) | Idiopathic NSIP |
| Bronchiolocentric interstitial pneumonia (BIP) | Secondary BIP (e.g., HP >>> CTD, aspiration, inhalational exposures, medications) | Idiopathic BIP (provisional diagnosis) |
| Diffuse alveolar damage (DAD) | Secondary DAD (multiple causes) | Idiopathic DAD (acute interstitial pneumonia) |
| Pleuroparenchymal fibroelastosis (PPFE) | Secondary PPFE (e.g., IPF, CTD, HP, medications, radiation, transplant [restrictive allograft syndrome], pulmonary infection [post-tuberculosis], occupational) | Idiopathic PPFE |
| Lymphoid interstitial pneumonia (LIP) | Secondary LIP (e.g., CTD, immune deficiency) | Idiopathic LIP |
| Alveolar filling patterns | ||
| Organizing pneumonia (OP) | Secondary OP (e.g., CTD, post-infectious, medications, aspiration) | Cryptogenic OP (idiopathic OP) |
| Respiratory bronchiolitis-ILD (RB-ILD) | Secondary RB-ILD (e.g., smoking >>> CTD, medications, aspiration, hereditary) | Idiopathic RB-ILD |
| Alveolar macrophage pneumonia (AMP) | Secondary AMP (e.g., smoking >>> CTD, medications, aspiration, hereditary) | Idiopathic AMP |
| Rare alveolar filling disorders | Acute & chronic eosinophilic pneumonia, pulmonary alveolar proteinosis, lipoid pneumonia | |
| Others | ||
| Combined pattern | Multiple combinations (e.g., NSIP + OP, UIP + PPFE) | |
| Unclassifiable pattern | Unclassifiable ILD (multiple undefined patterns) | |
Table 2.
UIP, usual interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; NSIP, nonspecific interstitial pneumonia; CTD, connective tissue disease; GGO, ground glass opacity; BIP, bronchiolocentric interstitial pneumonia; HP, hypersensitivity pneumonitis; DAD, diffuse alveolar damage; CTD-ILD, connective tissue disease-associated interstitial lung disease; PPFE, pleuroparenchymal fibroelastosis; LIP, lymphoid interstitial pneumonia; OP, organizing pneumonia; COP, cryptogenic organizing pneumonia; RB-ILD, respiratory bronchiolitis-interstitial lung disease; AMP, alveolar macrophage pneumonia.
Table 3.
|
Radiological-histological patterns |
Common differential diagnosis | |
|---|---|---|
| Dominant | Co-existinga | |
| UIP | NSIP | IPF, CTD-ILD, HP |
| BIP | HP, CTD-ILD, coexistent airway injury of other cause | |
| PPFE | Combined IPF and PPFE, CTD-ILD | |
| DAD | Acute exacerbation of IPF or other secondary UIP (e.g., CTD-ILD) | |
| AMP | IPF + smoking-related ILD | |
| NSIP | UIP | IPF, CTD-ILD, HP |
| OP | CTD-ILD, post-DAD, inhalation injury, HP, drug reaction | |
| BIP | HP, CTD-ILD | |
| LIP | CTD-ILD, drug-induced, post-viral (HIV, EBV) | |
| PPFE | Idiopathic PPFE, CTD-ILD | |
| BIP | OP | CTD-ILD, HP |
| UIP | HP | |
| NSIP | HP, CTD-ILD | |
| LIP | CTD-ILD | |
| PPFE | Idiopathic PPFE | |
| OP | NSIP | Post-infection, CTD-ILD, inhalation injury, HP, drug reaction |
| UIP | CTD-ILD, acute exacerbation of IPF | |
| BIP | HP, CTD-ILD | |
UIP, usual interstitial pneumonia; NSIP, nonspecific interstitial pneumonia; IPF, idiopathic pulmonary fibrosis; CTD-ILD, connective tissue disease-associated interstitial lung disease; HP, hypersensitivity pneumonitis; BIP, bronchiolocentric interstitial pneumonia; PPFE, pleuroparenchymal fibroelastosis; DAD, diffuse alveolar damage; AMP, alveolar macrophage pneumonia; ILD, interstitial lung disease; OP, organizing pneumonia; LIP, lymphoid interstitial pneumonia; HIV, human immunodeficiency virus; EBV, Epstein-Barr virus.
- References
- References
REFERENCES
1. Ryerson CJ, Adegunsoye A, Piciucchi S, et al. Update of the international multidisciplinary classification of the interstitial pneumonias: an ERS/ATS statement. Eur Respir J 2025;66:2500158.
[PubMed]2. American Thoracic Society; European Respiratory Society. American Thoracic Society/European Respiratory Society international multidisciplinary consensus classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2002;165:277–304.
[Article] [PubMed]3. Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–748.
[Article] [PubMed] [PMC]4. Wolters PJ, Blackwell TS, Eickelberg O, et al. Time for a change: is idiopathic pulmonary fibrosis still idiopathic and only fibrotic? Lancet Respir Med 2018;6:154–160.
[Article] [PubMed] [PMC]5. Raghu G, Remy-Jardin M, Richeldi L, et al. Idiopathic pulmonary fibrosis (an update) and progressive pulmonary fibrosis in adults: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2022;205:e18–e47.
[PubMed]6. Marinescu DC, Hague CJ, Muller NL, et al. Integration and application of radiologic patterns from clinical practice guidelines on idiopathic pulmonary fibrosis and fibrotic hypersensitivity pneumonitis. Chest 2023;164:1466–1475.
[Article]7. Churg A, Wright JL, Ryerson CJ. Pathologic separation of chronic hypersensitivity pneumonitis from fibrotic connective tissue disease-associated interstitial lung disease. Am J Surg Pathol 2017;41:1403–1409.
[Article] [PubMed]8. Liebow A, Carrington CB. The interstitial pneumonias. In: Simon M, Potchen EJ, LeMay M, eds. Frontiers of pulmonary radiology. New York: Grune & Stratton, 1969:102-141.9. Raghu G, Remy-Jardin M, Ryerson CJ, et al. Diagnosis of hypersensitivity pneumonitis in adults. An official ATS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med 2020;202:e36–e69.
[PubMed] [PMC]10. García-Manso IG, Arenas-Jiménez J, García-Sevila R, et al. Mosaic attenuation in non-fibrotic areas as a predictor of non-usual interstitial pneumonia pathologic diagnosis. Sci Rep 2022;12:7289.
[PubMed] [PMC]11. Ichikado K, Suga M, Muranaka H, et al. Prediction of prognosis for acute respiratory distress syndrome with thin-section CT: validation in 44 cases. Radiology 2006;238:321–329.
[Article] [PubMed]12. Ichikado K, Johkoh T, Ikezoe J, et al. Acute interstitial pneumonia: high-resolution CT findings correlated with pathology. AJR Am J Roentgenol 1997;168:333–338.
[Article] [PubMed]13. Collard HR, Ryerson CJ, Corte TJ, et al. Acute exacerbation of idiopathic pulmonary fibrosis. An international working group report. Am J Respir Crit Care Med 2016;194:265–275.
[PubMed]14. Craig PJ, Wells AU, Doffman S, et al. Desquamative interstitial pneumonia, respiratory bronchiolitis and their relationship to smoking. Histopathology 2004;45:275–282.
[Article] [PubMed]15. Hellemons ME, Moor CC, von der Thüsen J, et al. Desquamative interstitial pneumonia: a systematic review of its features and outcomes. Eur Respir Rev 2020;29:190181.
[Article]16. Lee JH, Chae EJ, Song JS, Kim M, Song JW. Pleuroparenchymal fibroelastosis in Korean patients: clinico-radiologic-pathologic features and 2-year follow-up. Korean J Intern Med 2021;36 Suppl 1:S132–S141.
[Article]17. Waseda Y, Johkoh T, Egashira R, et al. Antisynthetase syndrome: pulmonary computed tomography findings of adult patients with antibodies to aminoacyl-tRNA synthetases. Eur J Radiol 2016;85:1421–1426.
[Article] [PubMed]18. Flaherty KR, Travis WD, Colby TV, et al. Histopathologic variability in usual and nonspecific interstitial pneumonias. Am J Respir Crit Care Med 2001;164:1722–1727.
[Article] [PubMed]19. Guler SA, Ellison K, Algamdi M, Collard HR, Ryerson CJ. Heterogeneity in unclassifiable interstitial lung disease. A systematic review and meta-analysis. Ann Am Thorac Soc 2018;15:854–863.
[Article] [PubMed]20. Ryerson CJ, Urbania TH, Richeldi L, et al. Prevalence and prognosis of unclassifiable interstitial lung disease. Eur Respir J 2013;42:750–757.
[Article] [PubMed]21. Ryerson CJ, Corte TJ, Lee JS, et al. A standardized diagnostic ontology for fibrotic interstitial lung disease. An international working group perspective. Am J Respir Crit Care Med 2017;196:1249–1254.
[Article] [PubMed] [PMC]22. Noth I, Zhang Y, Ma SF, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013;1:309–317.
[Article] [PubMed] [PMC]23. Peljto AL, Selman M, Kim DS, et al. The MUC5B promoter polymorphism is associated with idiopathic pulmonary fibrosis in a Mexican cohort but is rare among Asian ancestries. Chest 2015;147:460–464.
[Article] [PubMed] [PMC]24. Newton CA, Oldham JM, Ley B, et al. Telomere length and genetic variant associations with interstitial lung disease progression and survival. Eur Respir J 2019;53:1801641.
[Article] [PubMed] [PMC]25. Choe EJ, Jang JH, Park JH, Jung SY, Her M, Lee JH. Role of blood Krebs von Lungen-6 in predicting acute exacerbation in patients with idiopathic pulmonary fibrosis. PLoS One 2025;20:e0323784.
[Article] [PubMed] [PMC]26. Song JW, Do KH, Jang SJ, Colby TV, Han S, Kim DS. Blood biomarkers MMP-7 and SP-A: predictors of outcome in idiopathic pulmonary fibrosis. Chest 2013;143:1422–1429.
[Article] [PubMed]27. Yokoyama A, Kondo K, Nakajima M, et al. Prognostic value of circulating KL-6 in idiopathic pulmonary fibrosis. Respirology 2006;11:164–168.
[Article] [PubMed]