


서 론
간질성 폐질환(interstitial lung disease, ILD)은 폐실질을 침범하는 비감염성, 비종양성 질환으로서 폐포 사이 공간인 간질(interstitium)에 세포의 증식과 분화, 만성적인 염증 및 섬유화 등 다양한 병리 기전이 따로 혹은 동시에 나타나는 질환군을 통칭한다[1]. 이 중 간질의 섬유화가 가장 흔한 표현형(phenotype)으로, 이로 인하여 환자는 폐활량의 감소 및 가스 교환의 장애를 경험하며 심할 경우는 호흡 부전 및 사망까지 이르게 된다. 약물, 유기 혹은 무기물질에 대한 노출, 방사선 치료, 결체조직질환 등 유발 원인을 알 수 있는 경우가 있고, 자세한 검사에서도 발병 원인이 밝혀지지 않을 때도 있는데, 이러한 경우를 특발성 간질성 폐렴(idiopathic interstitial pneumonia, IIP)이라 지칭하며 그중 가장 예후가 불량한 것이 특발성 폐섬유증(idiopathic pulmonary fibrosis, IPF)이다[2]. IPF는 65세 이상의 노인에서 호발하고, 수개월에서 수년에 걸쳐 진행하는 호흡곤란, 마른 기침 및 청진시 양측 폐하부의 흡기 수포음을 특징으로 하며, 환자들의 중간 생존기간은 진단 후 3-5년으로 알려져 있다[3]. 흡연은 IPF의 잘 알려져 있는 위험인자로, 그 밖에 위-식도역류, Epstein Barr virus나 Hepatitis C virus 등의 감염 및 유전자 변이 등도 질병 발생과 관련 있는 것으로 알려져 있다.
본 론
진단 과정
IPF가 의심되는 환자에서 병력 청취와 이학적 검사를 통하여 직업성 폐질환이나 결체조직질환 등 원인 감별이 가능한 질환 여부를 판단한다. 고해상도 흉부 전산화단층촬영(high resolution computed tomography, HRCT)을 시행하고, 이후 다학제 토론(multidisciplinary discussion, MDD)을 통하여 기관지폐포세척 검사(bronchoalveolar lavage, BAL)나 외과적 폐생검(surgical lung biopsy, SLB) 등의 추가적인 검사 시행 여부를 결정한다. 특징적인 IPF의 HRCT 소견을 보이는 경우(통상형 간질성 폐렴[usual interstitial pneumonia, UIP]형)는 조직 검사 없이도 IPF를 진단할 수 있으나, 그렇지 않을 경우는 조직 검사 등 추가적인 검사 후 다시 MDD를 통하여 최종적으로 IPF 진단을 하게 된다(Fig. 1).
병력 청취 및 기본 검사
고령자(특히 60세 이상의 남자)가 6개월 이상 진행되는 호흡곤란이나 마른 기침을 호소하고, 신체 진찰에서 곤봉지(clubbing) 혹은 흡기시 수포음이 들리거나, 흉부 X-ray에서 양폐하부의 증가된 음영을 보이는 경우 IPF를 의심해야 한다. 나이가 40-60세로 비교적 젊은 경우에도 폐섬유증의 가족력이 있다면 IPF의 가능성을 염두에 두고 자세한 병력 청취가 필요하다. 흡연력과 직업성 폐질환의 배제를 위하여 직업력에 대한 자세한 문진을 시행하고, 과민성폐장염 등의 가능성을 고려하여 환경에 대한 정보(거주하거나 근무하는 곳에서 곰팡이나 새와 장기간 접촉하였는지 등)를 얻어야 한다. 또한, 복용하는 약물이나 방사선 치료 여부 등에 대해서도 확인하여야 한다. 결체조직질환(connective tissue disease)의 동반 가능성을 고려하여 관절 및 피부 등 관련 증상에 대한 병력 청취가 필요하고, 관절 등 폐 외 증상 없이 ILD가 선행하는 경우가 있으므로 무증상이라도 항핵항체(antinuclear antibody)나 류마티스 인자(rheumatoid factor) 등 자가항체 검사를 시행한다[3].
고해상도 흉부 전산화단층촬영(HRCT)
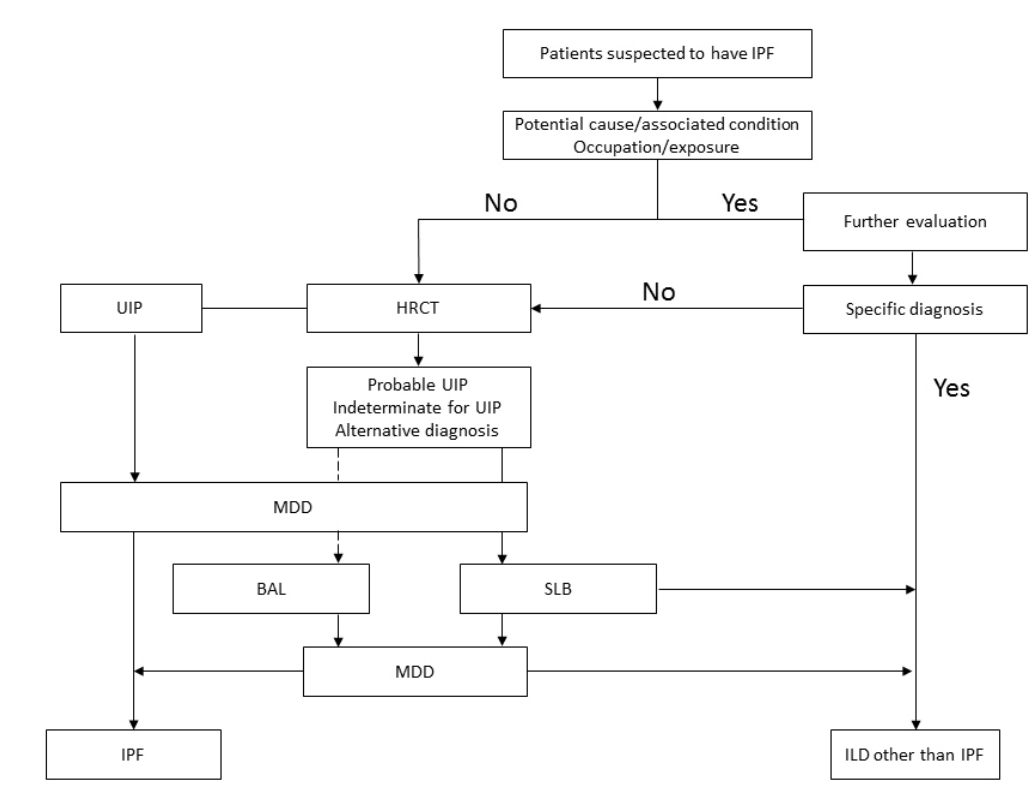
HRCT는 2 mm 이하의 얇은 두께(thickeness)를 갖는 영상을 1-2 cm 간격으로 촬영하여 폐실질의 변화를 민감하게 확인할 수 있는 CT 촬영기법이다[10]. 공기걸림(air trapping)이나 폐허탈(atelectasis) 등의 동반 여부를 확인하기 위하여 흡기 말 영상 외에 호기 말과 복와위 자세(prone position)에서도 촬영하는 것이 다른 ILD나 가성병소(pseudolesion) 감별에 도움이 된다[3] (Fig. 2).
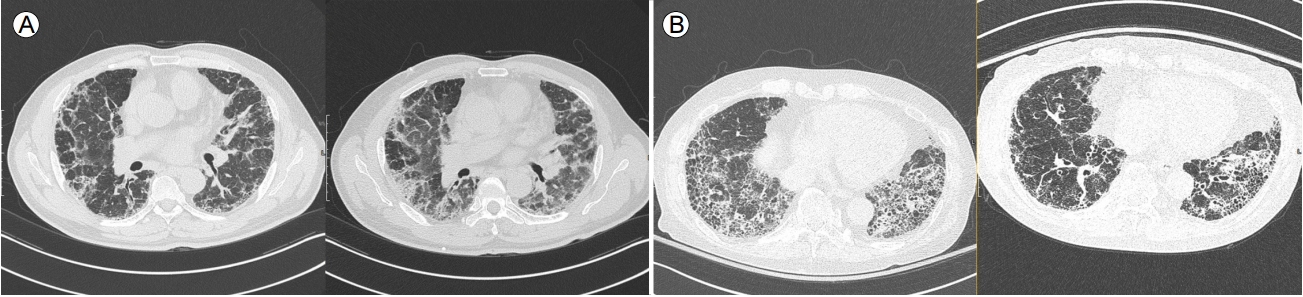
2011년에 발표된 IPF 진료 지침은 철저한 병력 청취, 자가항체 등의 검사에서 원인이 밝혀지지 않은 IIP 환자에서 HRCT를 먼저 시행하도록 하고, 그 소견에 따라 3가지 유형(UIP pattern, possible UIP pattern, inconsistent UIP pattern)으로 분류하여, UIP pattern인 경우를 제외하고는 SLB를 하도록 권고하였다[5]. HRCT 상 폐하부, 변연부 우세성으로 봉와양폐(honeycombing)와 망상음영(reticular abnormality)을 동반하고, 다른 질환을 시사하는 소견(섬유화 병변보다 범위가 더 넓은 간유리음영[ground glass opacity], 폐경결[consolidation], 중-상엽 침범 등)이 없는 경우, 영상의학적으로 UIP pattern으로 분류하고, 이러한 경우 병리 소견상 UIP pattern과 일치도가 매우 높은 것으로 보고되었다[11]. 기존의 진료 지침에서는 앞서살펴본 UIP pattern의 4가지 특징(폐하부/변연부 우세성, 봉와양폐, 망상음영, 다른 질환을 시사하는 소견이 없음) 중 봉와양폐를 제외한 나머지 3가지를 만족한 경우를 possible UIP pattern으로 정의하였는데, Chung 등[12]은 possible UIP pattern을 섬유화의 정도로 나누어 probable UIP (폐하부, 변연부 우세성, 망상음영 존재, 다른 질환을 시사하는 소견이 없음)와 indeterminate UIP pattern (CT 상 폐섬유화가 존재하나 definite, probable 혹은 inconsistent with UIP로 분류할 정도로 특징적이지 않은 경우)으로 나누어 병리조직 결과를 비교하였고, probable UIP pattern에서 병리 검사상 UIP pattern이 더 자주 관찰됨을 보고하였다(82.4% vs. 54.2%; p= 0.01). 이에 따라 2018년 개정된 지침에서는 흉부 HRCT 소견을 UIP, probable UIP, indeterminate for UIP, alternative diagnosis의 4가지로 분류하였다(Table 1, Fig. 3) [3,8].
외과적 폐생검(SLB)
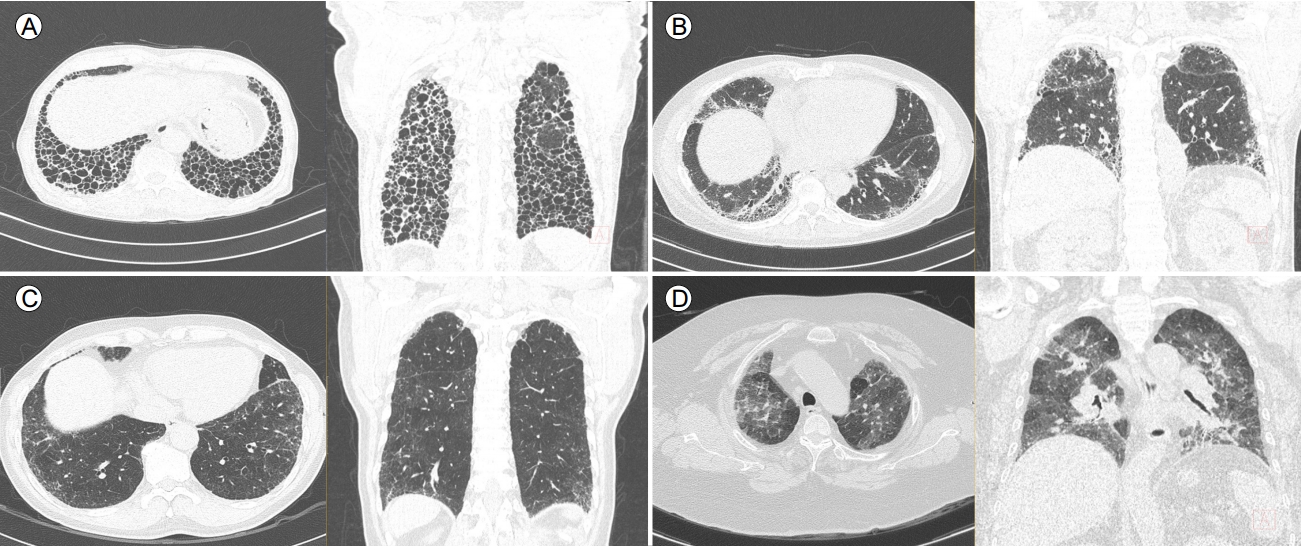
2018년 개정된 진료 지침에 따르면 조직 검사 소견도 영상과 마찬가지로 UIP, probable UIP, indeterminate for UIP, alternative diagnosis의 4가지로 분류하였다(Table 2, Fig. 4). 같은 환자의 서로 다른 두 엽에서 다른 조직형이 관찰될 수 있기 때문에(discordant UIP; 즉 한 엽에서는 UIP pattern, 다른 엽에서는 nonspecific interstitial pneumonia pattern), SLB 시행 시 최소 2곳 이상의 폐엽에서 조직을 얻어야 한다[3].
폐생검 후 발생할 수 있는 합병증으로는 급성악화(6.1%), 지속적인 공기 누출(6-12%), 감염(6.5%), 출혈(0.2-0.8%) 등이 알려져 있다[13]. Hutchinson 등[14]은 11년간 32,022명의 SLB를 받은 환자를 대상으로 사망과 연관된 인자를 분석하였고 남자, 고령, 기저 질환이 많을수록 사망이 유의하게 증가함을 보고하였다. 특히 75세 이상은 45세 미만에 비하여 4.5배 사망 위험도가 증가하였다. 또 안정시 혹은 운동시 산소를 필요로 하는 경우는 수술 후 사망, 폐렴, 지속적인 공기 누출 등의 주요 합병증이 발생할 확률이 증가한다. LoCicero [15]의 연구에서도 폐생검시 산소 요구량이 없을 경우 사망률은 4%, 수술 전 산소가 필요한 경우는 6% 그리고 호흡부전으로 기계 환기가 필요한 환자에서는 75%로 보고하였다. 조직 검사를 할 당시 급성악화의 병력도 알려진 위험인자로서, Park 등[16]은 SLB를 시행받은 200명을 대상으로 한 연구에서 급성악화시 조직 검사를 받은 경우 사망률(28.6%)이 그렇지 않은 경우(3.0%)에 비하여 유의하게 증가하고, 폐확산능(diffusing capacity for carbon monoxide)이 낮을수록 조직 검사 후 합병증이 발생할 위험이 높음(odds ratio, 0.959; 95% confidence interval, 0.930-0.989; p= 0.007)을 보여주었다. 그 외에 고령, 면역저하 상태, 중증의 폐고혈압이 있을 경우 수술 후 사망률이 증가하여 주의가 필요하다[15,17-19].
2011년 진료 지침에서 SLB에 대해서 비디오 흉강경 수술(video assisted thoracic surgery, VATS)과 개흉술(open lung thoracotomy, OLB)을 같은 수준으로 소개하였으나, 개정된 진료 지침에서는 VATS를 추천하였다. 41명을 대상으로 VATS와 OLB의 효용성을 비교한 연구에서, VATS 시행시 수술 시간(45.3 ± 12.2분 vs. 55.6 ± 11.2분)과 입원 기간(5.5 ± 1.3일 vs. 7.1 ± 2.3일)이 OLB에 비하여 줄어들었음을 보여주었고[20], 사망률도 유의하게 낮았다(1.18% vs. 2.29%, p< 0.001) [14].
최근 기관지 내시경을 이용한 냉동폐생검(bronchoscopic lung cryobiopsy) 시술이 ILD의 진단을 위한 조직 검사 방법으로 시도되고 있다[21,22]. 117명의 ILD 환자를 대상으로 냉동폐생검과 VATS를 비교한 연구에서, 조직의 적합성(100% vs. 100%)이나 ILD의 진단율(91% vs. 98%, p= 0.71) 및 시술 후 사망률(1.7% vs. 3.4%)이 두 방법 간 유의한 차이가 없는 것으로 보고되었다[22]. 하지만 각 기관별로 숙련도가 다르고, 시술 과정이 표준화되어 있지 않아 개정된 진료 지침에서는 아직 ILD를 진단하는 방법으로 권고하고 있지 않다[3].
외과적 폐생검(SLB)의 적응증
2011 American Thoracic Society (ATS)/European Respiratory Society (ERS)/Japanese Respiratory Society (JRS)/Latin American Thoracic Society (ALAT) 진료 지침과 동일하게 2018년 지침에서도 HRCT 상 전형적인 UIP형을 보이지 않는 경우는 SLB를 고려하도록 하였다. 그러나 2011년 지침이 발표된 이후 HRCT와 조직 소견의 일치율에 관한 연구들이 진행되었고, 흉부 HRCT 상 봉와양폐가 없더라도 흉막하에 망상음영과 견인성 기관지확장증을 동반한 경우 조직 검사에서 UIP가 진단될 확률이 높음(82-94%)이 확인되었다[11,12]. 특히 60세 이상 남자, 흡연력 등의 임상양상을 만족하는 경우에는 일치도가 더 높아짐을 근거로[23], 2018년 Fleishner 그룹에서는 IPF의 임상상을 보이는 환자에서 흉부 CT 상 probable UIP형을 보이는 경우 SLB 없이도 IPF를 진단할 수 있다고 제시하였다[8]. 하지만 이어 발표된 2018년 ATS/ERS/JRS/ALAT 진료 지침에서는 흉부 CT 검사 상 probable UIP 소견을 보이는 경우 SLB를 시행하도록 권유하였다(conditional recommendation) [3]. 비록 표면적으로는 흉부 CT 상 probable UIP 소견을 보이는 환자에 대해서 두 진료 지침의 견해가 다른 것처럼 보이지만, ATS/ERS/JRS/ALAT 진료 지침은 conditional recommendation으로서, HRCT에서 probable UIP 소견을 보이는 환자들의 다수(≥ 50%)는 SLB가 필요하지만 일부(<50%) 환자(예를 들면 임상적으로 IPF일 가능성이 높은)에서는 SLB가 필요하지 않을 수 있다라고 해석할 수 있다는 점에서 두 지침이 의도하는 바는 동일하다고 할 수 있다[24,25].
기관지폐포세척 검사
기관지폐포세척 검사(BAL)는 기관지 내시경을 이용하여 기도내 총 100-300 mL의 생리식염수를 3-5회 나누어 주입 후 회수하여 말초 기도와 폐포에서 검체를 얻는 검사방법이다[26]. 회수된 세척액은 주입된 양의 최소 30% 이상이 되어야 하며, 그 이하일 경우 세포 분율(differential counts)을 해석하는데 주의해야 한다.
IPF의 환자에서 BAL 세척액 소견은 정상인 혹은 다른 폐질환에 비하여 호중구의 비율이 증가하지만, 결체조직관련 폐질환이나 다른 질환에서도 유사한 소견을 보일 수 있어, 2011년 진료 지침에서는 IPF 진단을 위하여 BAL을 시행하도록 권고하지 않았다[5]. 그러나 IPF가 아닌 다른 질환을 감별하는데 도움이 될 수 있는데, BAL 세포분획에서 호산구가 25% 이상이면 호산구성 폐렴(eosinophilic pneumonia)에 진단적이고[26], 유육종증(sarcoidosis)의 경우 보통 림프구가 15% 이상이고 CD4/CD8 비율이 4 이상으로 높다. 따라서 흉부 CT에서 UIP pattern이 아닌 경우 상기 질환 등을 감별할 목적으로 BAL 세포분획과 CD4/CD8 비율을 확인하는 것이 권고 사항으로 새롭게 추가가 되었다[3].
다학제 진단의 중요성
IPF 진단의 표준은 다학제 토론을 통한 진단이다[3]. Flaherty 등[27]은 IIP의 진단 과정에서 임상, 병리, 영상 정보의 통합과 각 분야 전문가 간 토론을 통하여 진단의 일치도(interobserver agreement)와 신뢰도를 높일 수 있음을 보여주었다. Chaudhuri 등[28]은 지역 병원의 호흡기 전문의로부터 MDD를 의뢰받은 318명의 ILD 증례를 분석한 연구에서, 최초 진단이 IPF였던 107명 중 절반의 환자에서 MDD 후 진단이 변경되었으며, IPF를 제외한 다른 ILD 환자 136명도 33%에서 진단이 변경되었음을 보고하였다. Jo 등[29]도 호흡기 전문의로부터 의뢰받은 90명의 ILD 환자 중 MDD를 통하여 53%에서 진단이 변경되었고, 치료 방침에 있어서도 항섬유화제의 사용(3% vs. 21%, p< 0.01)이나 산소 치료(6% vs. 10%, p= 0.046) 등에 있어 유의한 변화가 있음을 보고하였다. 이러한 결과들은 MDD가 임상 진료에 있어서, 진단뿐만 아니라 환자의 치료 방침을 결정하고 예후를 판단하는 데에도 중요한 영향을 줌을 시사해 준다.
2018년 개정된 진료 지침에서는 MDD의 중요성을 더욱 강조하여 흉부 HRCT 정보를 얻은 후 MDD를 시행하여 BAL이나 SLB 등 추가적인 검사 시행 여부를 결정하도록 하였고, 상기 검사 결과들을 토대로 다시 한번 MDD를 할 것을 권유하였다(Fig. 1). 하지만 전형적인 IPF의 임상-영상상을 보이는 경우에는 반드시 MDD가 필요하지는 않다[3]. Fleischner 그룹도 조직 검사 시행 여부를 결정하거나, 조직 검사 후 임상/영상/병리 소견을 리뷰할 때, 질병의 경과가 알려진 것과 달라 재평가가 필요할 경우에 MDD를 추천하였다[8]. MDD의 구성은 경험이 많은 임상, 영상, 병리전문가를 기본으로, 필요시 환경, 유전, 류마티스질환 전문가가 참여하는 것이 권고된다. 하지만 참여 인원을 어떻게 구성할 것인지, 그리고 MDD 시행 주기 등에 대한 결정은 각 센터의 진료 여건에 맞게 시행될 수 있다.
결 론
IPF는 진단 후 지속적으로 진행하여 높은 사망률을 보이는 질환으로, IPF가 의심되는 환자의 조기 발견과 정확한 진단 및 적절한 치료가 매우 중요하다. 개정된 IPF 진단 지침은 흉부 HRCT 소견상 UIP형을 보이는 경우 외에도, 임상적으로 IPF가 의심되는 환자(working diagnosis of IPF)에서 SLB 없이도 IPF를 진단할 수 있는 여지를 두었다. 이러한 의사결정은 MDD를 통하여 진행되어야 하며, 이를 통하여 침습적 진단 방법이 필요한 환자를 선별하여 불필요하게 발생할 수 있는 수술 관련 합병증을 줄이고 정확한 진단을 내리는데 기여할 것으로 예상한다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






