


INTRODUCTION
Neurofibromatosis type 1 (NF1) is a genetic condition that occurs in 1 per 2,500-3,000 individuals, which makes it the most common autosomal dominant disorder in humans [1]. NF1 is a multisystemic progressive disease that affects the neurological, cardiovascular, gastrointestinal, endocrine, and orthopedic systems. Additionally, patients with NF1 have a higher overall risk of malignancy compared with the general population. This disease is caused by germline mutations of the NF1 gene, which maps to chromosome 17q11.2, spans approximately 350 kb genomic DNA, and contains 60 exons [2]. The NF1 gene encodes a protein named neurofibromin, which is composed of 2,818 amino acids and is expressed as a tumor suppressor in many cells [1]. The detection of mutations in NF1 is complicated due to its large size, the presence of pseudogenes, and the variety of mutations. If an individual has a mutation, penetrance approaches 100% by age 20. However, the expressivity of the mutated gene is highly variable, even among family members with the same mutation [3]. The present study is a case report of two siblings with NF1 who carried the same genetic mutation but had different clinical manifestations.
CASE REPORT
A 51-year-old woman presented at our endocrinology department with abnormal findings on a thyroid function test. At her regular medical check-up, the patient had thyroid stimulating hormone (TSH) and free thyroxine (T4) levels of 0.04 μIU/mL (reference value: 0.17-4.05 μIU/mL) and > 6.15 ng/dL (reference value 0.85-1.86 ng/dL), respectively. The patient, who was 150 cm tall and 57.3 kg in body weight, had been diagnosed with NF1 in her fourth decade but had no complaints of thyrotoxic symptoms such as hyperactivity, palpitation, heat intolerance, or fatigue. Her medical history included a tumor resection for a benign tumor in her right breast. Her blood pressure was 134/80 mmHg, her heart rate was 108 bpm, and her body temperature was 36.7°C. The patient’s thyroid gland was diffusely enlarged without tenderness, but there was no palpable nodule or mass. She had not been diagnosed with a developmental disorder during childhood and did not present with any spinal deformities or facial malformations. An ophthalmological examination revealed Lisch nodules (Fig. 1A) but no evidence of optic glioma. Additionally, the patient had multiple café-au-lait spots and skin neurofibromas (Fig. 1B), as well as skin fold freckling (Fig. 1C), but no plexiform neurofibromas.
A diagnosis of NF1 is made if any two of the following seven criteria are met: (a) two or more neurofibromas on or under the skin or one plexiform neurofibroma, (b) freckling of the groin or the axilla (armpit), (c) six or more café-au-lait spots measuring 5 mm at the greatest diameter in prepubescent individuals and > 15 mm at the greatest diameter in post-pubescent individuals, (d) skeletal abnormalities such as sphenoid dysplasia or a thinning of the cortex of the long bones of the body, (e) two or more Lisch nodules (hamartomas of the iris), (f) optic glioma, and/or (g) a first-degree relative with NF1 [4]. The patient had multiple skin neurofibromas, freckling on the groin and axilla, multiple café-au-lait spots, and several iris Lisch nodules, which satisfied four of the seven criteria. Furthermore, her family history suggested autosomal dominant inheritance of NF1, because her mother and her mother’s siblings had multiple skin neurofibromas, and her younger brother was diagnosed with NF1 in our hospital.
An evaluation of the patient’s thyrotoxicosis and her thyroid scan revealed the hot uptake of both thyroid lobes (Fig. 2), and her TSH receptor antibody level was 18.1 IU/L (upper limit value is 1.75 IU/L). She was diagnosed with Graves’ disease and treated with propylthiouracil (PTU; 300 mg per day) and propranolol (60 mg per day) in divided doses. Six weeks later, the patient’s thyroid function had improved, and her free T4 value decreased to 1.43 ng/dL, but her aminotransferase value was more than three-fold the normal upper limit. Thus, the PTU treatment was discontinued due to possible hepatotoxicity, which is a major side effect of this therapy. The patient agreed to radioiodine therapy, and I-131 16 mCi was administered. Six weeks later, her liver function results had normalized, but her free T4 level was 0.81 ng/dL and her TSH level 42.88 µIU/mL, indicating hypothyroidism. The patient began taking levothyroxine (25 µg/day) and now enjoys well-being in a euthyroid state. An abdominal computed tomography (CT) scan revealed a 1.5 cm mass in her left adrenal gland with 50 Hounsfield units (HU) (Fig. 3), but a hormonal analysis showed that it was a non-functioning adenoma. Annual CT scans and hormonal analyses showed no significant changes in the size or hormonal activity of the tumor.
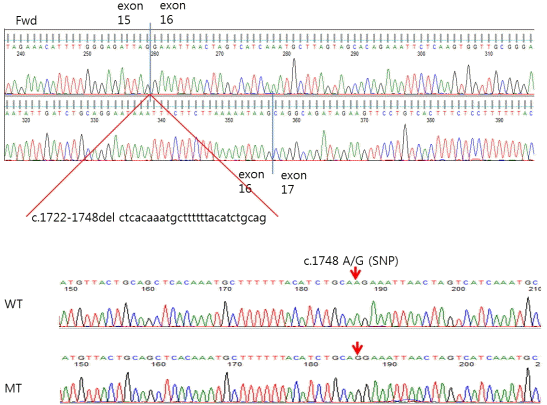
As mentioned above, the present patient had a family history of NF1; her younger brother was diagnosed with NF1 in our hospital, and her mother and all of her mother’s siblings had multiple neurofibromas. Furthermore, the patient’s cousin had multiple skin neurofibromas, which suggests that she was also an NF1 patient (Fig. 4). The patient’s genomic DNA was isolated from her peripheral white blood cells using the Flexigene DNA kit (Qiagen, Hilden, Germany), and polymerase chain reaction (PCR) was performed using intronic primers flanking the 60 exons of the NF1 gene. The purified PCR products were sequenced using the ABI3730 3 1 genetic analyzer (Applied Biosystems, Foster City, CA, USA) and analyzed using FinchTV (Version 1.2.0, Geospiza, Seattle, WA, USA). All nucleotide numbers refer to the wild-type genomic DNA sequence of the NF1 gene as logged at the National Center for Biotechnology Information. The analyses revealed a c1748A > G mutation in exon 16 and loss of 27 base pairs (bp) (c. 1722_1748del) in the NF1 genes of both the patient and her brother (Fig. 5). All laboratory and genetic investigations were conducted with informed consent from the proband, and the research protocol was approved by Ethics Committee of Incheon St. Mary’s Hospital.
DISCUSSION
NF1 is a heritable autosomal dominant genetic syndrome caused by heterogeneous mutations in the NF1 gene, which is a large gene that contains 57 constitutive exons and spans approximately 350 kb genomic DNA [2]. Ko et al. [5] conducted a study on 60 Korean NF1 families and found only seven recurrent mutations, which implies that there is a widespread distribution of NF1 mutations. Furthermore, genotype-phenotype analyses have indicated the lack of a clear relationship between specific NF1 mutations and the clinical features of the disease. Ko et al. [5] reported that skin manifestations (café-au-lait spots were observed in all patients, whereas freckling and cutaneous neurofibromas were observed in 70-80% of patients) and Lisch nodules were the most common phenotypes, followed by bone lesions (39.7%), intracranial lesions (30.8%), and malignancy (7.7%). Previous studies have also reported coexistent non-Hodgkin's lymphoma, intra-abdominal malignant peripheral nerve sheath tumors, and malignant gastrointestinal stromal tumors in Korean patients with NF1 [6,7].
The present patient and her brother exhibited the nucleotide change c.1748A > G, which generated a cryptic splice site and caused the loss of 27 bp from the mRNA transcript (r.1722-1748 del). This mutation has previously been reported by Brinckmann et al. [8] but has yet to be identified in a Korean patient. Despites carrying the same genetic mutation, the patient and her brother exhibited different clinical symptoms other than the typical skin manifestations, which is consistent with previous studies. The present patient had iris Lisch nodules, adrenal incidentaloma, and Graves’ disease, but it is unclear if her Graves’ disease was associated with NF1. Several cases of NF1 patients presenting with Graves’ disease have been published. For example, Sakane et al. [9] reported the case of a 35-year-old female NF1 patient who was referred due to hypertension, tachycardia, and diagnosed Graves’ disease, and Bolko et al. [10] described the case of a 42-year-old female patient with NF1 who also had hyperthyroidism and diagnosed Graves’ disease.
An important issue is that the thyrotoxic symptoms and signs of these two diseases, including hypertension, tachycardia, and excess sweating, are similar to those of pheochromocytoma (PHEO), which is caused by excessive levels of catecholamines. Nonetheless, a high degree of suspicion by clinicians is necessary to avoid overlooking thyroid function abnormalities in NF1 patients who show thyrotoxic manifestations, because PHEO has a higher incidence in NF1 patients compared with the general population and because hyperthyroidism is not commonly associated with NF1. The younger brother of the present patient had a milder form of NF1 relative to his sister; he had only skin neurofibromas and freckling on the groin and axilla. Further molecular studies are needed to better understand the mutations of the NF1 gene and its variable expression patterns, as well as to anticipate specific complications associated with these mutations.
The present study describes two patients in a single NF1 family. Genetic analyses revealed that they had the same 27-bp deletion mutation in the NF1 gene that resulted in c. 1748A > G in exon 16. However, the patients exhibited different clinical manifestations. Thus, further study is necessary to reveal the molecular processes underlying gene expression and phenotypes. A better understanding of the genetics of NF1 will allow clinicians to diagnose NF1 more accurately, even in patients who are suspected of having this disease but do not fulfill the diagnostic criteria. It will also aid in the earlier detection of clinical complications and provide improved genetic counseling to NF1 families.




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






