거인증의 감별진단을 통해 확인된 클라인펠터 증후군 1예
A Case of Klinefelter Syndrome Differentially Diagnosed as a Cause of Gigantism
Article information
Abstract
클라인펠터 증후군은 잉여의 X염색체 존재에 의한 선천성 질환으로 남성의 일차성 성선기능저하증을 유발하는 가장 흔한 원인의 하나이다. 이는 고환 위축, 불임 등의 성선기능저하증과 관련된 증상 이외에도 여러 다양한 합병증을 동반하는 것으로 잘 알려져 있다. 저자들은 군 복무 중인 환자에서 거인증에 대한 감별진단을 통해 확진된 클라인펠터 증후군 환자를 경험하여 이를 보고하고자 한다.
20세 남자 환자가 지속적으로 키가 커지는 증상으로 내원하였다. 환자의 신장은 193.4 cm로 거인증과 관련된 다른 원인들에 대한 감별진단을 시행하였으나 모두 정상이었다. 하지만 환자의 신체 검사상 이차 성징의 증거가 뚜렷하지 않았고 호르몬 검사상 일차성 성선기능저하증이 의심되었다. 이에 염색체 핵형 검사를 시행하여 47, XXY의 클라인펠터 증후군으로 확진되었다.
Trans Abstract
Klinefelter syndrome is congenital disease that is associated with the existence of an extra X chromosome, and is one of the most common causes of male primary hypogonadism. In addition to hypogonadism-associated manifestations such as testicular atrophy and infertility, it is also well known that this syndrome may be associated with other systemic comorbidities. In this report, we describe a typical case of Klinefelter syndrome that was differentially diagnosed as a cause of gigantism. A 20-year-old male was admitted to evaluate the cause of tall stature. His height was 193.4cm, and al screening tests for gigantism were negative. Physical examination revealed no clear evidence of secondary sexual characteristics, and the results of a hormonal assay were highly suspicious for primary hypogonadism. Based on these findings, we performed a chromosomal analysis and confirmed Klinefelter syndrome with a 47, XXY Karyotype. (Korean J Med 2011;80:343-347)
서 론
클라인펠터 증후군은 잉여의 X염색체 존재에 의한 선천성 질환으로 남성의 일차성 성선기능저하증을 유발하는 가장 흔한 원인의 하나이다[1,2]. 이는 고환 위축, 불임 등 성선기능저하증과 관련된 증상 이외에도 폐질환, 악성 종양 및 내당능장애 등 다양한 합병증을 동반하는 것으로 잘 알려져 있다[3-7].
하지만, 복잡한 임상양상과 이 질병에 대한 전문적 이해의 부족으로 인해 상당수의 클라인펠터 증후군 환자들에게서 정확한 진단과 치료가 지연되고 있으며 이로 인해 많은 환자들이 다양한 합병증으로 인한 삶의 질 저하를 경험하고 있다[2,8].
최근 저자들은 군 복무 기간 중 거인증에 대한 감별진단을 통해 확진된 전형적인 클라인펠터 증후군 환자를 경험하였으며 이를 보고하고자 한다.
증 례
환 자: 20세 남자
주 소: 지속적으로 키가 커지는 증상
현병력: 군 복무 중이던 환자는 입대 전 신장이 190 cm였으나 입대 후에도 지속적으로 키가 자라 군 복무 11개월 간 약 3 cm의 신장 증가를 경험하였다. 내원 6개월 전 환자는 신장 증가에 대해 외부 병원에서 선별검사를 시행하였으나 당시 측정한 영상 검사 및 혈청 IGF-1 376.6 ng/mL (연령에 대한 참고치 219~644 ng/mL)로 정상 판정을 받았으며 이후 특별한 검사 또는 처치 없이 지내고 있었다.
신체 검사: 신장 193.4 cm, 체중 75 kg이었고, 입원 당시 혈압 120/80 mmHg, 심박수 66회/분, 체온 36.5℃로 활력 지수는 정상이었다. 시진상 환자는 상체에 비해 하체의 길이가 길었다(Fig. 1A and 1B). 또한 여성형 유방이 관찰되었으며 음낭과 성기의 크기가 현저하게 작고 액모와 치모가 거의 관찰되지 않았다(Fig. 1C and 1D). 환자의 2차 성징 발현 정도는 Tanner stage II에 해당하였다.
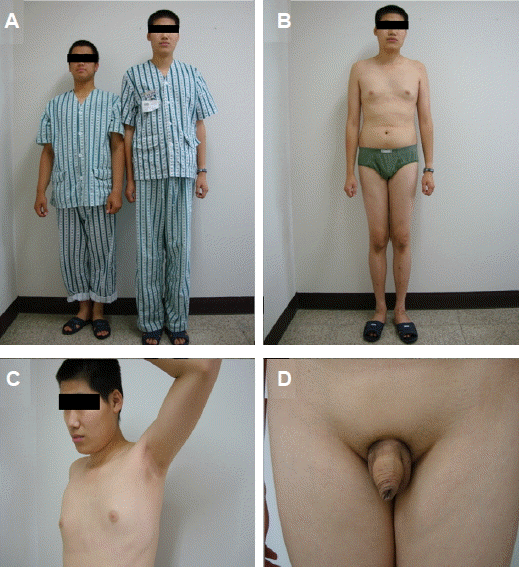
Physical examination findings of the patient. (A) Tall stature (193 cm) compared with that of normal subject (173 cm). (B) Increased leg and arm lengths compared with the upper part of the body. (C) No axillary hair with gynecomastia. (D) Small penis and scrotum with scarce pubic hair.
과거력: 특이 사항 확인되지 않았다.
가족력: 환자는 1남 1녀 중 둘째로 환자의 부모 및 형제 중 거인증과 관련된 병력을 가진 사람은 존재하지 않았다.
혈액 검사 결과: 일반혈액검사, 생화학 검사, 그리고 소변검사상 환자에게 특별한 이상은 관찰되지 않았다. 경구 당부하검사 결과에서도 유의한 내당능장애 또는 당뇨병이 의심되는 증거는 확인되지 않았다. 하지만 내분비 검사상 혈청 LH 11.49 mIU/mL, FSH 36.54 mIU/mL로 현저하게 증가되었으며 혈청 테스토스테론 0.57 pg/mL, 24시간 소변 유리 테스토스테론 0.56 ng/mL로 유의하게 감소되어 있었다(Table 1). 이외 다른 뇌하수체 기저 호르몬 검사 결과 모두 참고치 이내에 해당하였다.

Laboratory findings of the patient
영상 검사 결과: 뇌하수체 자기공명 영상 결과 뇌하수체에 좌우엽간 비대칭이 존재하고 우측 뇌하수체에 경계가 명확하지 않은 저음영의 영역이 존재하여 임상적으로 비기능성 뇌하수체 미세선종이 의심되었다(Fig. 2A and 2B). 음낭 초음파 검사상 환자의 양측 고환에 유의한 저형성 변화(hypoplasia) 및 내부에 다수의 고음영 결절이 관찰되었다(Fig. 2C and 2D). 하지만 종양 또는 국소 병변의 증거는 관찰되지 않았다. 3차원 측정을 통하여 계산된 양측 고환의 부피는 우측 0.99 cc, 좌측 0.82 cc (참고치 15~30 cc) 감소되어 있었다.
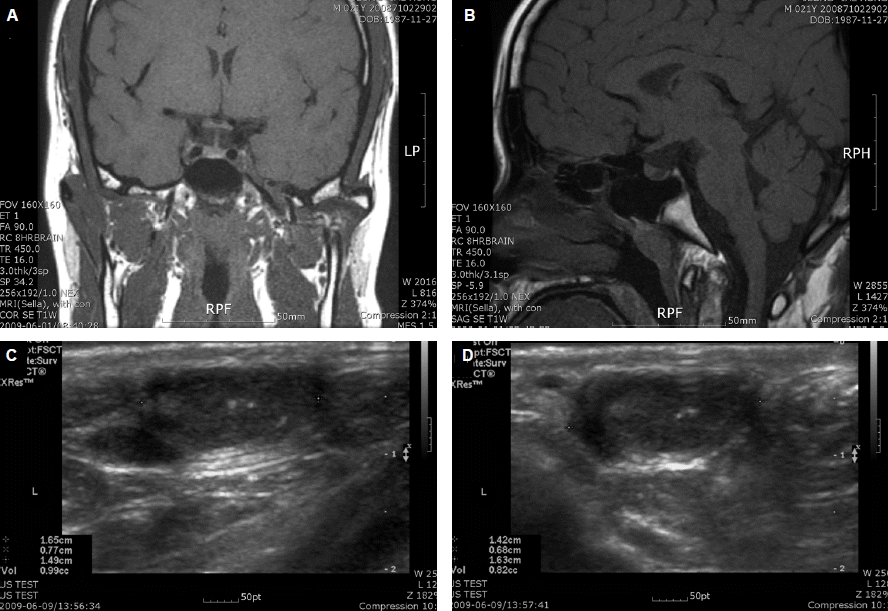
Radiologic findings of the patient. (A, B) Ill-defined lesion with low signal intensity was observed in pituitary gland of the patient. (C, D) Significantly small testes volume (right 0.99 cc, left 0.82 cc) observed on testicular ultrasonography.
치료 및 경과: 환자는 뇌하수체 선종 및 Marfan 증후군 등 거인증의 원인이 될 수 있는 다른 원인들에 대한 감별진단을 시행하였으나 유의한 이상은 관찰되지 않았다. 기저 호르몬 검사 결과 뇌하수체 미세선종을 시사하는 임상적 근거 역시 존재하지 않았다. 하지만 신체 검사상 환자는 2차 성징이 지연되어 있었고, 음낭 초음파 검사 결과 양측 고환의 크기가 현저하게 감소되어 있었다. 또한 호르몬 검사상 LH와 FSH의 유의한 상승, 테스토스테론의 유의한 감소가 확인되었다. 이에 환자의 신장 증가의 원인으로 일차성 성선기능저하증을 의심하여 염색체 핵형 검사(karyotyping)와 정액 검사를 추가로 시행하였다. 검사 결과 환자의 핵형은 47, XXY로 확인되었으며 정액 검사는 환자의 정액량이 너무 적어 실패하였다(Fig. 3). 검사 결과에 따라 환자는 클라인펠터 증후군으로 확진되었으며 곧 전역하였다. 현재 환자는 민간 의료기관에서 테스토스테론 대체요법으로 치료하며 경과를 관찰하고 있다.

Chromosomal analysis revealed a karyotype of 47, XXY, which is typical for Klinefelter syndrome.
고 찰
클라인펠터 증후군은 일차성 성선기능저하증을 유발하는 가장 흔한 원인의 하나로 남아 1,000명 당 약 1명의 비율로 발견되는 것으로 알려져 있다[1,2]. 일반적으로 염색체 핵형 검사를 통해 진단이 이루어지며 하나 이상의 잉여 X염색체가 관찰되는 것이 특징적인 검사 소견이다[9]. 환자들의 대다수가 47, XXY 유전자형을 보이지만 48 XXXY, 49XXXXY, 46, XY/46XXY 모자이크 현상(mosaicism) 등 다양한 유전자형을 보이는 환자들이 보고되어 있다[9-13]. 클라인펠터 증후군의 염색체 이상은 감수분열 과정 중 한쪽 부모의 성염색체가 분리되지 않아 일어나는 것으로 알려져 있으며 모자이크 현상이 관찰되는 환자들은 수정 후 유사분열 시기에 일어난 염색체 비분리현상에 의한 것으로 알려져 있다[9]. 잉여의 X염색체 수가 늘어날수록 환자의 임상 증상이 심해지는 것으로 알려져 있으며 안드로겐 수용체 유전자의 CAGn 다형성 길이와 환자의 증상간에도 상관관계가 있는 것으로도 알려져 있다[10,14].
클라인펠터 증후군의 성선 기능 저하와 관련된 증상으로 세정관 및 Leydig 세포의 손상과 고환의 위축이 관찰되며, 정자 수 감소, 불임, 혈청 FSH와 LH 농도 상승, 혈청 테스토스테론 농도 감소 및 2차 성징의 지연 등이 알려져 있다[9,15]. 성선 외 증상으로는 장골(long bone) 이상에 의한 하지 길이 이상이 생길 수 있고 판단력 저하, 학습 능력 저하 등에 의해 사회 관계를 형성하는데 어려움을 경험한다[16,17]. 이외에도 만성 기관지염, 기관지 확장증, 폐기종 등의 호흡기 질환과 생식 세포 종양, 유방암, 비 호지킨 림프종 등의 악성 종양, 하지 정맥류, 자가면역 질환 그리고 내당능 장애 및 당뇨병 등을 동반한 환자들의 증례가 보고되어 있다[3-7,18].
하지만 이렇게 다양한 임상양상으로 인한 진단의 어려움과 이 질병에 대한 의료진의 전문적 이해 부족으로 현재 많은 사람들에게서 정확한 진단이나 치료가 지연되고 있는 것으로 추정된다. 일부 연구들에 의하면 사춘기 이전에 클라인펠터 증후군으로 진단되는 경우는 전체 환자의 10% 미만으로 추정되며 초기 성인기에 이르러서도 약 26%의 환자만이 진단되는 것으로 알려져 있다[2,8]. 우리나라의 경우 관련된 역학 연구가 충분하지 못한 실정이므로 그 실태를 정확하게 파악할 수는 없지만 87명의 비교적 많은 환자들을 대상으로 시행된 조 등의 연구에서 대상자들의 진단 당시 평균 연령 26세에 달한다고 보고한 바 있어 외국의 상황과 크게 다르지 않을 것으로 판단된다[13]. 본 증례에서 환자는 193.4 cm에 달하는 신장 증가를 주소로 내원하여 뇌하수체 선종, Marfan 증후군 등 거인증을 유발할 수 있는 다른 질병들에 대한 감별 진단을 통해 클라인펠터 증후군으로 확진되었다. 이 환자 역시 성선기능 저하증 외에는 비교적 양호한 임상경과를 보이는 환자임에도 과거에 내원한 다른 병원들에서는 정확한 진단이 이루어지지 않아서 신장 증가와 같은 추가적인 합병증에 대한 관리가 제대로 이루어지지 못했던 것으로 판단된다.
클라인펠터 증후군에 있어 성선기능 저하증과 관련된 제증상은 테스토스테론 보충요법을 통해 치료할 수 있으며 일부의 환자들에 있어서는 인공 수정을 통해 성공적인 임신이 가능하다는 연구들도 보고되고 있다[9]. 이는 적절한 의학적 관리를 통해 클라인펠터 증후군 환자들의 합병증을 조기에 관리하고 삶의 질을 유의하게 향상시킬 수 있음을 의미한다. 현재 추정되는 클라인펠터 증후군의 유병률을 토대로 추산한다면 아직 상당수의 환자들에게서 정확한 진단과 치료가 지연되고 있을 가능성이 높다. 앞으로 이에 대한 임상가들의 지속적인 관심 제고가 필요할 것이다. 아울러 의료에 대한 접근성이 지속적으로 향상되고 유전자 카운슬링을 비롯하여 질병을 조기에 진단할 수 있는 다양한 진단기술이 지속적으로 발전됨에 따라 향후 클라인펠터 증후군 환자들에 대한 진단과 치료가 조기에 적극적으로 이루어지고 환자들의 임상 경과에 긍정적인 변화가 일어날 것으로 전망된다. 이와 관련된 좀 더 많은 연구들이 진행되어야 할 것으로 판단된다.