간을 침범한 유전성 출혈모세혈관확장증에서 발생한 ST 분절 상승 급성 심근경색증
ST Elevation Myocardial Infarction in a Patient with Hereditary Hemorrhagic Telangiectasia Involving the Liver
Article information
Trans Abstract
This is a case report of a 71-year-old woman with hereditary hemorrhagic telangiectasia (hereditary hemorrhagic telangiectasia [HHT], Osler–Weber–Rendu syndrome) involving the liver who developed ST elevation myocardial infarction and died from aggressive coronary thrombosis. HHT is an autosomal dominant hereditary disease associated with mutations of genes that regulate the endothelial surface. It has characteristic muco-cutaneous telangiectasia and other common manifestations are epistaxis, gastrointestinal bleeding, and iron-deficiency anemia. In addition, arteriovenous malformations or vascular ectases commonly occur in the pulmonary, hepatic, and cerebral circulations. Hemorrhages and thrombosis can both develop from these vascular abnormalities in HHT. Most thrombotic events are forms of venous thrombosis, such as deep vein thrombosis, while arterial thrombosis occurs infrequently. We present a case of aggressive coronary thrombosis in HHT, as a rare complication of HHT.
INTRODUCTION
Hereditary hemorrhagic telangiectasia (HHT), also known as Osler–Weber–Rendu disease, is a rare autosomal dominant hereditary disease. It is characterized by epistaxis, muco-cutaneous and visceral telangiectasia, and inheritance. Major complications of HHT include bleeding from the compromised vascular beds at the characteristic telangiectases or arteriovenous malformations (AVMs) and resulting anemia [1].
Paradoxically, thrombotic conditions are relatively common in patients with HHT. Deep vein thrombosis occurs frequently and it can lead to systemic embolic infarction via a right-to-left shunt through a pulmonary AVM. In comparison, arterial thrombosis, such as coronary thrombosis, is unusual in HHT.
We present a patient with HHT involving the liver who developed ST elevation myocardial infarction (STEMI) and subsequently died from aggressive coronary thrombosis.
CASE REPORT
A 71-year-old woman visited the hospital for evaluation of suspicious intrahepatic vascular dilatations found during abdominal ultrasonography for a medical checkup. She had no specific symptoms and her medical history was non-contributory. Laboratory tests showed low hemoglobin (6.5 g/dL), but normal platelet count and prothrombin and activated partial thromboplastin times. Additional laboratory tests for anemia showed decreased serum iron (10 μg/dL) and low total iron binding capacity (234 μg/dL), but normal ferritin (74.85 ng/mL). Liver computed tomography (CT) revealed prominent tortuous dilatations of the hepatic arteries and portal vein, suggestive of liver involvement of HHT (Fig. 1).
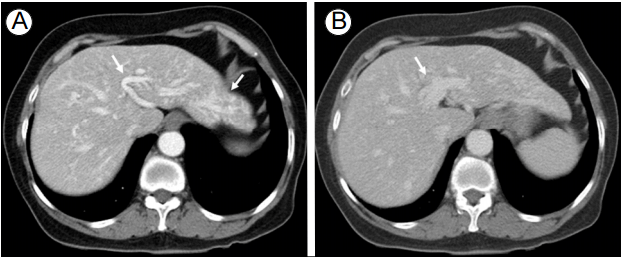
The dynamic liver computed tomography images. (A) The arterial phase shows tortuous dilatation of the hepatic artery (arrows), and (B) the portal phase shows tortuous dilatation of the portal vein (arrow), suggestive of hepatic involvement of hereditary hemorrhagic telangiectasia.
There were no telangiectases on her skin or oral mucosa. She had had frequent epistaxis since childhood. Her father, brother, two sisters, daughter, and nephew also had frequent epistaxis. Her brother had spontaneous brain hemorrhage at the age of 67 years. We recommended that she and her family undergo genetic testing for HHT mutations. However, only the patient could afford the tests financially. We tested ENG and ACVRL1 and identified an ACVRL1 mutation (Table 1) [2].

Results of ACVRL1 gene mutation
Twenty-eight days later, she visited the emergency room with fever, chills, nausea, and vomiting. She had slightly low blood pressure (105/55 mmHg), mild tachycardia (102/min), and fever (38-39℃); laboratory tests showed leukocytosis (11,480/mm3) and high C-reactive protein (CRP) level (23.23 mg/dL). Abdominopelvic CT was suggestive of two early hepatic abscesses (Fig. 2). She was immediately treated with intravenous (IV) antibiotics and fluids. Fever continued, but leukocytosis normalized and CRP level decreased during hospitalization.
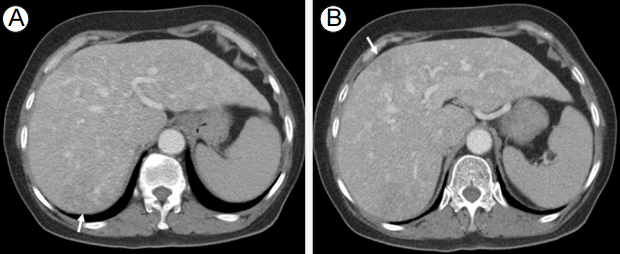
(A, B) The post-contrast phase of the abdominopelvic scan shows two newly developed fuzzy low-density areas in the right lobe of the liver (arrow), suggestive of early liver abscess formations.
On hospital day 4, she felt squeezing anterior chest pain associated with diaphoresis. Cardiac enzyme tests and an electrocardiogram (ECG) performed immediately showed elevated cardiac enzymes and ST segment elevation in leads II, III, and aVF, consistent with an inferior wall STEMI. We scheduled emergency coronary angiography. Considering the bleeding risk of HHT, we decided not to administer anti-platelet agents, but only IV heparin during the angiography.
The coronary angiography showed total occlusion of the proximal to middle right coronary artery with large thrombi. We performed balloon angioplasty and thrombi aspiration, but thrombi remained in the totally occluded distal right coronary artery (Fig. 3). Since reperfusion failed, we decided to administer IV abciximab and perform a second percutaneous coronary intervention. Although her vital signs stabilized and the chest pain resolved, the ECG still showed ST segment elevation and the cardiac enzymes were markedly elevated after the first coronary angiography. About 7 hours later, the second coronary angiography was performed and thrombi were seen in the distal right coronary artery. We repeated the thrombus aspiration, but cardiac arrest occurred during the procedure. The coronary angiography showed another thrombotic occlusion in the proximal left anterior descending artery (Fig. 4). Despite additional thrombus aspiration, the residual thrombi remained and the cardiogenic shock continued. We decided to stop the procedure, and start extracorporeal membrane oxygenation and continuous renal replacement therapy due to hypotension, bradycardia, and metabolic acidosis. We treated her intensively, but she died 5 hours after the coronary angiography.
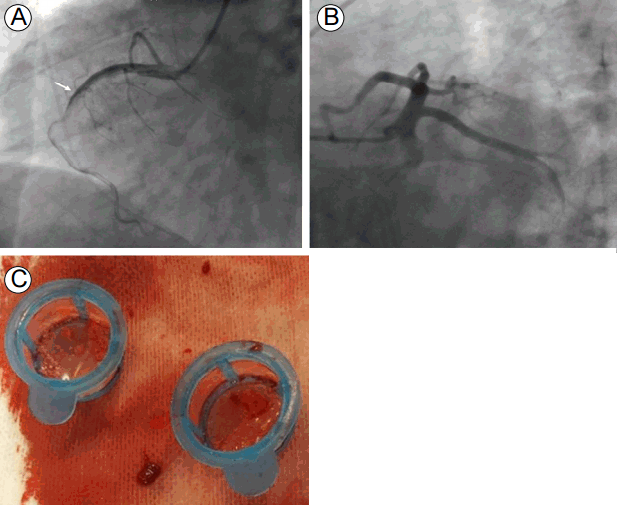
(A) The emergency coronary angiography shows thrombotic occlusion in the proximal to middle right coronary artery (arrow), and (B) the normal left anterior descending and circumflex arteries. (C) The thrombi aspirated from the right coronary artery.
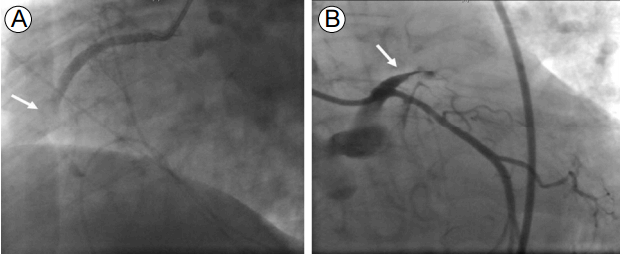
(A) The second coronary angiography shows thrombotic occlusion in the distal right coronary artery with remaining thrombi (arrow). (B) After cardiac arrest, coronary angiography shows another thrombotic occlusion in the proximal left anterior descending artery (arrow).
DISCUSSION
Hereditary hemorrhagic telangiectasia is caused by mutations in three major genes: ENG, protein product endoglin (HHT type 1, HHT1); ACVRL1, protein product activin receptor-like kinase 1 (HHT type 2, HHT2); and MADH4, protein product Smad4 (juvenile polyposis-HHT overlap syndrome). Over 80% of the patients with HHT will have mutations in either the ENG or ACVRL1 genes [3]. ACVRL1 mutations, which are associated with HHT2, are reported to have higher incidence of hepatic vascular abnormalities, while HHT1 has higher incidence of pulmonary vascular abnormalities [4].
Garcia-Tsao et al. [5] categorized complications of hepatic involvement of HHT into three main forms: high-output heart failure; portal hypertension; and biliary disease. There are no demonstrated associations between hepatic abscesses and HHT, but invisible biliary abnormalities might have contributed to the development of hepatic abscesses in this case.
Patients with HHT may be in an inherent prothrombotic state. A recent study found significantly elevated von Willebrand factor (vWF) and factor VIII (FVIII) levels in adults with HHT, and the degree of FVIII elevation correlates with future thrombotic risk. There is also correlation between low serum iron levels and thrombosis in patients with HHT [6]. The serum iron levels decrease because of bleeding, and the iron depletion leads to increased thrombosis to limit blood loss from injured vessels. This condition may have caused the elevated coagulation factors.
Arterial thrombosis, especially coronary thrombosis, is uncommon in HHT. There are some reports of myocardial infarction in patients with HHT, but most of these patients had a paradoxical embolism through a pulmonary AVM or coronary artery aneurysms [7]. Our patient was at low risk of acute coronary syndromes and there was no evidence of pulmonary AVMs or coronary aneurysms. This suggests that thrombotic complications of HHT can occur in arteries as well as in veins. Studies suggest that venous and arterial thrombosis are different phenomena of the same disease, and that the difference is dependent on individual predisposing factors or co-morbidities [8]. Furthermore, high vWF and FVIII levels seem to be risk factors for both arterial and venous thrombosis [9].
In summary, patients with HHT have high risks of thrombosis and hemorrhage, and the thrombotic complications of HHT can be critical, such as aggressive thrombosis in the coronary arteries. The FVIII, vWF, and serum iron levels should be determined to estimate the future thrombotic risks in patients with HHT. Further studies need to investigate the details of coronary thrombosis in HHT patients.