골수증식종양 진단의 발전
Advances in the Diagnosis of Myeloproliferative Neoplasms
Article information
Trans Abstract
Philadelphia chromosome-negative classical myeloproliferative neoplasms include polycythemia vera, essential thrombocythemia, and primary myelofibrosis. In recent years, there have been major advances in our understanding of the molecular pathophysiology of these rare subgroups of myeloproliferative neoplasms. The World Health Organization diagnostic criteria were revised in 2008, and incorporated new somatic mutations of JAK2 V617F, found in most patients with polycythemia vera, essential thrombocythemia, or primary myelofibrosis. Subsequently, other mutations (MPL W515 and CALR) were discovered and this led to substantial changes in the diagnosis and treatment guidelines. This article reviews the diagnostic criteria for Philadelphia chromosome-negative classical myeloproliferative neoplasms, and changes in the diagnostic algorithm for clinical practice in Korea.
서 론
골수증식종양(myeloproliferative neoplasms, MPN)은 하나 이상의 골수세포(과립구계, 적혈구계, 거대핵세포, 비만세포)가 과도하게 증식하는 클론성 조혈모세포 혈액질환으로, BCR-ABL1 양성 만성 골수백혈병(chronic myelogenous leukemia), 만성 호중구백혈병(chronic neutrophilic leukemia), 진성적혈구증가증(polycythemia vera), 본태혈소판증가증(essential thrombocythemia), 일차골수섬유증(primary myelofibrosis) 등이 포함되어 있다[1]. 발생 초기에는 조혈기능 증가로 인해 말초혈액 혈구와 골수세포충실도의 증가를 보이지만, 단계적으로 진행하여 말기에는 골수섬유화, 비효과조혈(ineffective hematopoiesis) 또는 급성모세포기(acute blast phase)로 진행하여 골수부전에 이르게 된다.
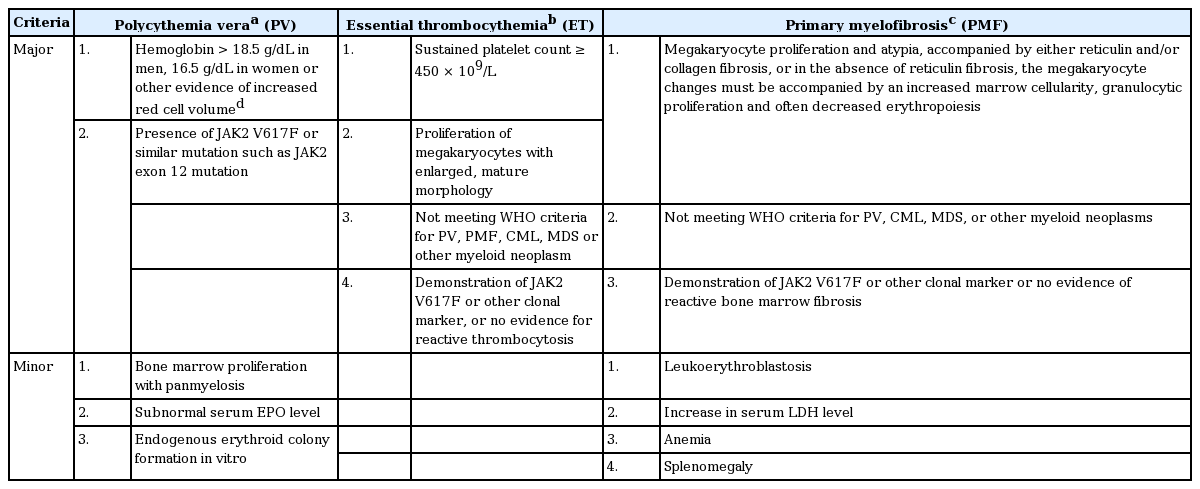
만성 골수백혈병을 제외한 진성적혈구증가증, 본태혈소판증가증 및 일차골수섬유증을 통상 ‘필라델피아염색체 음성’인 전형적인 골수증식종양(philadelphia chromosome-negative classical MPN)이라고 한다[2]. 만성 골수백혈병의 BCR-ABL1 유전자를 제외하고 다른 골수증식종양의 진단은 주로 임상양상을 통한 감별 진단과 추적검사 등의 비특이적인 방법으로 이루어져 왔으나, 2005년 Janus kinase 2 (JAK2) 티로신인산화효소의 V617F 돌연변이(JAK2 V617F)가 보고되면서 진단과 연구의 새로운 전환점을 맞이하게 되었다[3,4]. 따라서 2008년 World Health Organization (WHO) 분류에서도 임상적, 혈액학적 특성을 바탕으로 한 기존의 진단기준에 더하여 JAK2 유전자 및 MPL 유전자변이와 같은 분자유전학적 표지자가 진단에 필수적인 검사로 포함되었다(Table 1) [1].
최근에는 JAK2, MPL 유전자변이 외에도 CALR 유전자변이 등 새로운 분자진단 표지자와 검사법이 빠르게 발전하면서 질환의 병태생리를 이해하고 진단의 정확성을 높이는데 크게 기여하고 있다[5,6]. 국내 골수증식종양의 유병률은 지난 10년간 약 3.8배 증가하였으며[2], 국내 검사실의 혈액종양진단을 위한 표준화된 지침이 발표되는 등[7], 골수증식종양의 진단에 대한 관심이 높아지고 있으므로 이에 본 종설에서는 필라델피아염색체 음성 골수증식종양인 진성적혈구증가증, 본태혈소판증가증, 일차골수섬유증 진단의 발전에 대해 알아보고자 한다.
본 론
진성적혈구증가증
정상적인 적혈구 생성 조절 기전과는 무관하게 적혈구 생성이 증가되는 특징을 갖는 골수증식종양으로 거의 모든 환자에서 JAK2 V617F 돌연변이(> 95%) 또는 이와 기능적으로 유사한 JAK2 엑손 12번 돌연변이(~5%)가 발견되며, 적혈구계뿐 아니라 과립구계, 거대핵세포계도 함께 증식하는 범혈구증가증(panmyelosis)이 관찰된다[1]. 주요 임상증상으로는 적혈구용적 증가로 인한 고혈압이나 혈관 장애로, 약 20%의 환자에서 정맥 또는 동맥의 혈전증이 관찰된다. 두통, 어지러움, 시각장애, 감각이상 등의 증상을 흔히 동반하며, 그 외에도 소양증이나 홍색사지통증(erythromelagia), 통풍을 호소하는 경우도 흔하다[1]. 질환이 진행되면서 다혈색(plethora)과 비장비대(70%), 간비대(40%) 등의 소견을 동반한다[1].
진성적혈구증가증은 3단계로 진행되며, 적혈구증가 정도가 경미하거나(pre-polycythemic phase) 적혈구 부피가 크게 증가하는 시기(overt polycythemia)에는 골수세포충실도가 평균 80% 이상으로 연령대비 정상 범위에 비해 크게 증가하고, 말초혈액과 골수에서 범혈구증가증을 보인다. 질환이 진행하게 되어 적혈구증가-후기 골수섬유증(post-polycythemic myelofibrosis)에 이르면 비효과조혈을 동반한 혈구생성 감소와 함께 골수외 조혈에 의한 비장비대, 골수화생(myeloid metaplasia)으로 인해 말초혈액에서 백적모세포증(leukoerythroblastosis), 눈물방울 모양의 적혈구를 동반한 변형적혈구증가증(poikilocytosis) 소견이 관찰될 수 있다. 이 시기에 레티쿨린(reticulin) 또는 콜라겐(collagen) 면역조직화학염색을 통해 현저한 골수섬유화를 확인할 수 있으며, 골수세포충실도는 다양할 수 있지만 질환 초기와는 달리 연령대비 오히려 감소되어 있는 경우도 흔하다[1]. 2008 WHO 분류에 따른 진성적혈구증가증의 진단기준은 표 1에 제시된 바와 같다.
세포유전학적 이상은 진성적혈구증가증 진단시 약 20%에서 볼 수 있다. 가장 흔히 관찰되는 핵형이상은 +8, +9, del (20q), del (9p) 등으로 특히 적혈구증가-후기 골수섬유증 단계에 주로 나타나며, 골수형성이상이나 급성 골수성백혈병으로 이행할 경우 100%에서 세포유전학적 이상을 보인다[1].
본태혈소판증가증
일차적으로 거대핵세포계가 증식하는 골수증식종양으로 말초혈액에서 지속적인 450 × 109/L 이상의 혈소판증가증과 함께 골수에서 크고 성숙한 거대핵세포의 증가 소견이 관찰된다[1]. 본태혈소판증가증 환자들은 대부분 긴 무증상기를 보이나 일부에서는 혈전증이나 출혈경향을 나타낼 수 있다.
말초혈액에 증가된 혈소판은 크기가 다양하고, 드물게 위족(pseudopod) 또는 무과립혈소판과 같은 이상한 형태로 관찰될 수 있다. 골수조직검사에서 세포충실도는 정상 또는 중등도로 증가되어 있고, 특징적인 형태학적 소견으로 크기가 크고 세포질의 양이 풍부하며 핵은 깊게 패이고 과분엽화되어 마치 사슴뿔 모양처럼 보이는 성숙한 거대핵세포가 증식된 소견이 관찰된다[1]. 혈소판증가증은 초기 일차골수섬유증의 경우에도 동반될 수 있는데, 본태혈소판증가증에 비해 골수섬유화로 진행할 가능성이 높고 생존율이 더 낮으며 진단에 따른 치료 전략이 다르기 때문에 감별진단이 중요하다[8,9]. 기존 연구에 의하면, 본태혈소판증가증으로 진단받은 438명의 환자들을 WHO 기준으로 재분류하였을 때 162명의 실제 본태혈소판증가증 이외에 나머지는 일차골수섬유증의 전섬유화(184명)단계이거나 초기 섬유화(137명)단계였다는 보고가 있다[10]. 본태혈소판증가증은 특징적인 모양의 거대핵세포가 증가하고 호중구 및 적혈구 생성 증가나 좌방이동(left shift)이 없다. 이에 비해 일차골수섬유증은 비정상적인 형태의 비정형 거대핵세포가 관찰되고 과립구 증식에 따른 골수세포충실도증가와 적혈구 조혈감소가 동반되어 백적모세포증, 혈청 lactate dehydrogenase (LDH) 증가, 빈혈, 비장종대의 소견을 보이는 것이 주요 차이점이다[1].
드물게 환자의 일부에서는 진단 후 수년 뒤 골수섬유화와 골수화생, 골수외 조혈로 진행할 수 있으며, 2008 WHO 분류에 본태혈소판증가증-후기 골수섬유증(post-essential thrombocythemia myelofibrosis)에 대한 진단기준이 명시되어 있다. 이전에 본태혈소판증가증으로 진단된 환자에서 중등도 이상의 골수섬유화가 관찰되면서 빈혈, 백적모세포증, 비장비대의 증가, LDH의 증가, 임상증상의 악화(6개월간 10% 이상의 체중감소, 야간발한, 37.5도 이상의 이유를 알 수 없는 발열 중 하나 이상의 기준 만족) 중 2개 이상 만족시 진단이 가능하다[1,11].
분자유전학적 이상으로 JAK2 V617F 돌연변이가 환자의 40-50%에서 검출되며, JAK2 V617F 변이가 검출되지 않는 환자 중 1%에서는 MPL 유전자의 W515 돌연변이가 검출된다[1]. 2008 WHO 분류에 따른 본태혈소판증가증의 진단기준은 표 1에 제시된 바와 같다.
일차골수섬유증
일차골수섬유증은 골수의 거대핵세포와 과립구계의 뚜렷한 증식과 함께 골수외 조혈을 동반한 심한 골수섬유화를 특징으로 하는 골수증식종양이다[1]. 환자의 약 30%는 진단시 무증상이거나 또는 건강검진에서 비장비대 소견이나 혈액 검사에서 빈혈, 백혈구 증가, 혈소판 증가가 우연히 발견되어 진단되는 경우가 있다. 주요 증상은 피로감, 호흡곤란, 체중감소, 야간발한, 미열, 출혈경향 등을 보이며, 골수외 조혈에 의한 비장비대(90%)와 간비대(50%) 동반이 흔하다.
특징적인 혈액학적 소견으로는 말초혈액에 눈물방울모양의 적혈구를 동반한 대소부동변형적혈구증가증(anisopoikilocytosis)과 함께 미성숙골수전구세포 및 백적모세포증이 관찰되고 혈청내 LDH가 증가한다. 골수는 세포충실도가 높으면서 심한 레티쿨린 또는 콜라겐섬유화를 보이는데, 골수섬유화 정도는 질환이 진행되면서 점차 심해진다. 일차골수섬유증의 전섬유화 또는 초기 섬유화단계(pre-fibrotic and early stage)는 골수세포충실도가 높은데 이 시기에 주로 증식된 세포는 호중구와 비정형 거대핵세포이다. 특히 비정형 거대핵세포는 심한 비정상 형태를 취하며 골수 혈관동과 뼈잔기둥 주위에 집단적으로 분포하고 grade 0-1 정도의 섬유화를 나타낸다. 질환이 진행되어 섬유증단계(fibrotic stage)가 되면 미성숙 조혈세포가 군데군데 모여있으나, 골수모세포는 10% 미만이다. 이 시기에도 비정형 거대핵세포가 큰 집단을 이루는 특징을 보인다. 골수섬유화는 grade 2-3 정도의 심한 섬유화가 관찰되고 이와 더불어 혈관이 증식되고, 골경화(osteosclerosis)도 동반될 수 있다[1]. 가장 흔히 골수외 조혈이 일어나는 장기는 비장으로 주로 비장적수가 팽창되고 신생혈관 생성이 증가하며, 간에서도 골수외 조혈을 볼 수 있다.
JAK2 V617F변이는 일차골수섬유증 환자의 약 50%에서 나타나고, 기능적으로 유사한 MPL 유전자의 W515 돌연변이가 5%에서 검출된다[1]. 세포유전학적 이상은 진단시 약 30%에서 관찰되며, del (13) (q12-22), der (6) t (1;6) (q21-23; q21.3), del (20q), partial trisomy 1q 등을 볼 수 있다.
골수증식종양의 분자진단
골수증식종양의 진단을 위해서는 먼저 감염, 염증질환, 종양과 같은 기저질환에 의한 반응성혈구증가증이 배제되어야 하고, 만성 골수백혈병의 진단기준이 되는 BCR-ABL1 융합유전자가 검출되지 않아야 한다. 또한, 골수형성이상증후군이나 다른 골수증식종양에 해당하는 소견이 없어야 한다. 최근 골수증식종양진단에 유용한 분자유전학적 이상은 다음과 같다.
JAK2 유전자 돌연변이
JAK2 단백은 티로신인산화효소(tyrosine kinase)로 조혈성장인자수용체로부터 신호전달을 시작하는데 중요한 역할을 담당한다. JAK2 유전자의 617번째 코돈인 발린(valine)에서 페닐알라닌(phenylalanine)으로 바뀌게 되는 JAK2 V617F 돌연변이는 기능획득 돌연변이로 JAK2 단백의 구조적 활성화를 유도하여 성장인자와 무관하게 지속적으로 골수구계 세포의 증식을 일으키는 것이 밝혀졌다[3,4,12]. 이후 많은 임상연구를 통하여 JAK2 V617F 돌연변이가 필라델피아염색체 음성 골수증식종양에서 가장 흔히 발견되는 유전자변이이며, 보고에 따라 다소 차이가 있지만 진성적혈구증가증의 90-95%, 본태혈소판증가증의 35-70%, 일차골수섬유증의 50% 정도에서 검출되는 것이 확인되었다[13]. JAK2 V617F 돌연변이는 진성적혈구증가증, 본태혈소판증가증, 일차골수섬유증 외에도 비만세포증, 만성 호중구백혈병 등 다른 골수증식종양과 만성 골수단구성백혈병, 골수형성이상증후군과 같은 골수증식종양이 아닌 다른 골수질환에서도 낮은 빈도이지만 보고가 되고 있어 질환특이적인 표지자는 아니지만[14], 각 환자마다 검출되는 JAK2 V617F 유전자변이의 양이 서로 다르고 이에 따라 질환의 감별 진단 및 예후 예측에 유용하다고 알려져 있다[15,16]. JAK2 V617 돌연변이가 검출되지 않은 진성적혈구증가증 환자의 일부에서는 JAK2 유전자의 엑손 12번에서 기능적으로 유사한 체세포 돌연변이가 검출된다[17].
MPL W515 돌연변이
MPL 단백은 트롬보포이에틴 신호전달에 관여하는 세포막수용체(thrombopoietin receptor)로서 거대핵세포의 분화와 혈소판 증식을 조절하는데 관여한다. MPL 유전자 엑손 10번에 위치한 5개의 아미노산 모티브(RWQFP motif, Arg-Trp-Gln-Phe-Pro)는 MPL 단백의 자발적 활성화를 억제하는데 중요한 역할을 담당하는데, 515번째 코돈인 트립토판(tryptophan)이 다른 아미노산으로 치환되는 MPL W515 기능획득돌연변이가 발생하면 MPL 단백의 구조적 활성화를 유도하여 지속적인 신호전달을 유발한다[18]. 보고에 따라 다소 차이가 있지만, 본태혈소판증가증의 1%, 일차골수섬유증의 5%에서 MPL W515 돌연변이가 검출되며[18], 진성적혈구증가증에서는 현재까지 단 2예만이 보고되었다[19]. MPL W515돌연변이 양성군은 돌연변이 음성군에 비해 혈색소와 총 백혈구수가 낮고 높은 혈소판수를 보인다고 알려져 있다[20,21]. 국내 연구 결과에 따르면, 본태혈소판증가증 환자의 3%에서 MPL W515 돌연변이가 관찰되었고 JAK2 V617F 음성인 환자 중에서는 7-12%의 비율로 확인되었다[22,23]. 외국의 보고와 마찬가지로 MPL W515 돌연변이 양성 환자들에서 MPL W515 돌연변이 음성 또는 JAK2 V617F 양성 환자에 비해 총 백혈구수와 혈색소가 유의하게 낮았다고 보고된 바 있다[22]. MPL W515 돌연변이의 아형은 트립토판이 류신(leucine)으로 치환되는 W515L과 라이신(lysine)으로 치환되는 W515K가 가장 흔하며, 그 외에도 아르지닌(arginine)으로 치환되는 W515R, 알라닌(alanine)으로 치환되는 W515A 등이 드물게 보고되고 있다[24].
CALR 유전자 돌연변이
최근 발표된 Calreticulin (CALR) 유전자 엑손 9번 돌연변이는 골수증식종양에서 JAK2 유전자 돌연변이 다음으로 흔하게 발견되는 분자유전학적 이상소견으로, 전체 본태혈소판증가증 환자의 25%, 일차섬유골수증환자의 35%에서 발견되지만 진성적혈구증가증 환자에서는 발견되지 않는다[5,6]. CALR 단백은 다기능 소포체단백(multifunctional endoplasmic reticulum)으로, 단백 폴딩 및 칼슘조절을 비롯한 통합적 기능을 수행한다[25]. CALR 유전자 돌연변이로 인해 칼슘조절기능에 중요한 4개의 중요한 아미노산 모티브(KDEL motif, Lys-Asp-Glu-Leu)가 소실되는 것이 확인되었지만, 아직까지 골수증식종양의 발병기전은 명확히 밝혀지지 않았다[5,6,25]. 이제까지 보고된 CALR 유전자 돌연변이 중 52개의 염기가 결실되면서 틀이동변이(frameshift)가 발생하는 type 1 돌연변이가 50%로 가장 흔하고, 그 다음으로 type 2 돌연변이(5개의 염기 TTGTC가 삽입)가 30%를 차지하며, 그 외에도 약 40여종에 이르는 다양한 틀이동변이들이 보고되었다[5,6].
CALR 유전자 돌연변이가 검출되는 골수증식종양 환자들은 JAK2 또는 MPL 유전자 돌연변이를 보유하고 있는 환자에 비해 높은 혈소판수, 낮은 백혈구 및 헤모글로빈 수치를 보이나 혈전증의 위험이 낮고 상대적으로 양호한(indolent) 임상양상을 보이는 것으로 보고되었다[5,25]. 국내 연구 결과에 따르면, CALR 유전자 돌연변이는 본태혈소판증가증 환자의 18-25%, 일차골수섬유증 환자의 15-21%에서 관찰되었고[26-28], 외국의 보고와 마찬가지로 CALR 유전자 돌연변이 양성 환자들에서 JAK2 유전자 돌연변이 양성 환자에 비해 총 백혈구수와 혈전증의 위험이 낮다고 보고하였다[26,27]. CALR 유전자 돌연변이의 검출은 본태혈소판증가증, 일차골수섬유증의 진단 및 임상소견, 예후적 차이를 보이는 등 유용한 표지자로서 향후 개정될 WHO 분류에 도입될 수 있을 것으로 사료된다.
기타 유전자변이
일부 골수증식종양에서 TET2, EZH2, DNMT3A, ASXL1, IDH1/2 등의 유전자이상이 발견되지만[29], JAK2 또는 MPL 유전자 돌연변이와 동시에 나타날 수 있으며 골수증식종양 외 다른 혈액질환에서도 검출될 수 있어 아직까지 진단적인 유용성은 충분히 검증되지 않았다.
골수증식종양의 진단적 접근방법
최근 대한진단혈액학회에서는 국내 대학병원 및 종합병원을 대상으로 혈액종양진단을 위한 검사현황과 국제 혈액종양 진단 지침을 바탕으로 혈액종양 초기 진단 검사항목 지침을 수립하여 발표한 바 있다[7]. 골수증식종양 진단시 반드시 시행해야 할 필수 검사항목을 1차 검사로, 추천 검사항목은 2차 항목으로 정리되어 있으며, 감별 진단이 필요한 골수형성이상/골수증식종양(myelodysplastic/myeloproliferative neoplasms)에 속하는 질환도 함께 포함되어 있다(Table 2). 만성 골수백혈병의 진단기준이 되는 BCR-ABL1 융합유전자와 JAK2 V617F 검사는 우선적으로 시행해야 하는 필수 검사항목에 해당한다. JAK2 V617F 검사 결과 음성인 경우, 의심되는 질환에 따라 다음으로 시행해야 하는 분자 진단 검사항목이 다른데, 진성적혈구증가증일 경우 JAK2 엑손 12번을 검사하며, 본태혈소판증가증과 일차골수섬유증에서는 MPL W515와 CALR 엑손 9번을 검사하여 돌연변이 여부를 확인한다.

결 론
골수증식종양 진단기준은 계속해서 변화하는 분야이며, 특히 진단에 유용한 분자유전학적 이상이 보고됨에 따라 질환을 이해하고 진단의 정확성을 높이는데 기여하고 있다. 가장 잘 알려진 분자유전학적 이상인 JAK2 V617F 돌연변이 이외에도 MPL W515 돌연변이, CALR 유전자 돌연변이 등의 새로운 표지자가 골수증식종양 중 특히 진성적혈구증가증, 본태혈소판증가증, 일차골수섬유증에서 높은 검출률이 보고되고 있다. 분자유전학적 이상 여부를 검출하는 것은 진단뿐 아니라 임상소견의 차이 및 질환 예후의 추정 등에 있어 중요하며 나아가 임상에서 치료제 개발과 선택에도 고려되어야 할 중요한 의미를 가지므로 앞으로도 지속적으로 연구가 필요할 것으로 생각된다.