전신홍반루푸스로 오인된 혈관내B대세포림프종 1예
A Case of Intravascular Large B-cell Lymphoma Mimicking Systemic Lupus Erythematosus
Article information
Abstract
저자들은 1997년 ACR 분류기준 및 2012년 SLICC의 분류기준을 만족하여 SLE로 진단되었지만 적절한 치료 반응을 보이지 않아 추가적인 검사를 통해 IVLBCL로 진단된 환자의 증례를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
Trans Abstract
Intravascular large B-cell lymphoma (IVLBCL) is a rare subtype of non-Hodgkin’s lymphoma (NHL) and that progresses rapidly and is usually fatal. Because it usually presents with nonspecific symptoms, such as fever, the early diagnosis of IVLBCL is very difficult and it is often misdiagnosed as another disease. Systemic lupus erythematosus (SLE) is an autoimmune disease that affects various organs. The clinical manifestation of SLE ranges from rash and arthritis through anemia and thrombocytopenia to serositis, nephritis, seizures, and psychosis. Thus, it can be easily confused with many other disorders. We report a case of IVLBCL mimicking SLE in the initial diagnosis.
서 론
혈관내B대세포림프종(intravascular large B-cell lymphoma, IVLBCL)은 B세포 계열의 비호지킨림프종(non-Hodgkin’s lymphoma, NHL)의 드문 종류로 혈관내 악성 림프구의 증식을 특징으로 하는 질환이다. 초기에 발열, 피로감 등을 보이며 검사상 빈혈, 적혈구 침강계수의 상승, 젖산탈수소효소의 상승 등 비 특이적인 소견을 보여 진단이 어렵고 질병의 진행 속도가 빠르고 급격하기 때문에 짧은 기간에 환자가 사망에 이를 수 있으므로 조기 진단 및 감별 진단의 중요성이 강조되는 질환이다[1]. 전신홍반루푸스(systemic lupus erythematosus, SLE)는 자가항체형성을 통해 피부, 신장, 신경계, 근골격계, 심혈관, 폐, 조혈기관 등 전신을 침범하는 대표적인 자가면역질환이며, 침범 기관에 따라 그 임상양상 역시 피부 발진부터 단백뇨, 신경병증, 흉수, 빈혈, 혈소판감소증에 이르기까지 다양하게 나타난다. 또한 SLE는 특정 장기를 침범하는 다른 자가면역질환과는 달리 환자의 기저상태에 따라 발병시 나타나는 주 임상양상이 개개인마다 다양하며 이후에도 질병경과에서 따라 다양한 임상증상 및 소견을 보인다. 이러한 이유로 인해 SLE는 초기 진단시 오진되기 쉬워 다른 질환, 특히 악성종양과의 감별이 필요하다[2,3]. 본 저자들은 초기 진단에서 SLE로 오인된 IVLBCL의 증례를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
증 례
주 소: 발열과 오한
현병력: 60세 여자 환자가 3일 전 시작된 발열과 오한을 주소로 개인병원진료 후 항생제를 투약받았으나 증상의 호전을 보이지 않아 전원되었다.
과거력: 특이한 병력은 없었다. 내원 한 달 전 넘어지면서 왼쪽 옆구리를 부딪힌 외상력을 가지고 있었다.
가족력: 특이한 가족력은 없었다.
신체검사 소견: 내원시 활력 징후는 혈압 102/60 mmHg, 맥박 98회/분, 호흡 22회/분, 체온은 39.1℃였다. 급성병색을 보였고, 의식은 명료하였다. 안면부에 특이소견은 없었으나 구강경구개의 통증을 동반하지 않은 궤양소견이 관찰되었다. 흉부소견상 호흡음 및 심음은 정상적으로 청진되었다. 복부소견상 좌늑골하연으로 비장이 2횡지 정도 촉진되었고, 좌측이 조금 더 심한 양측의 늑골척추각 통증이 있었으나 압통은 저명하지 않았다. 환자는 입원 당시 3-4일 전부터 시작된 좌측 안면마비를 호소하였으며, 이에 대한 신경학적 검사상 양측 전정와우신경, 좌측 얼굴신경 마비 소견을 보였다.
검사실 소견: 말초혈액 검사에서 백혈구 2,580/mm3 (중성구 66.3%, 림프구 24.60%), 혈색소 10.0 g/dL, 헤마토크리트 26.1%, 혈소판 73,700/mm3로 범혈구 감소증 소견을 보이고 있었다. 혈청 생화학 검사상 AST 87 IU/L, ALT 56 IU/L, ALP 1,231 IU/L, LDH 2,905 IU/L, C-반응단백질(CRP) 5.35 mg/dL로 모두 상승 소견을 보였다. 페리틴(ferritn) 1,182.1 μg/L (정상치: 6-85 μg/L), 중성지방(triglyceride) 276 mg/dL (정상치: 45-150 mg/dL)의 증가도 관찰되었다. 소변검사상 단백뇨(+/-), leukocyte (1+), WBC (1-2)이었다. 내원시 흉수소견이 있어 진단적인 흉수 천자를 시행하였고 검사상 여출액이었으며 배양검사를 포함하여 특이소견이 관찰되지 않는 이차성 흉수소견을 보였다. 혈액 검사상 지속적인 범혈구 감소증 소견을 보여 원인평가를 위해 자가면역질환 및 용혈성 빈혈에 대한 검사를 시행하였고, 신경학적 이상소견에 대해서 뇌척수액 검사를 시행하였다. 자가면역질환 검사결과 류마티스 인자(RA factor) 정상(9 IU/mL, 정상치: 0-18 IU/mL), 항dsDNA항체 정상(2.4 IU/mL, 정상 0-30), 항Sm항체 음성, 항cardiolipin항체 IgG/IgM 정상(0.1/0.3, 정상치: < 23/< 11), 항beta2GP1항체 정상(0.1, 정상치: < 20) 소견을 보였으나, 보체의 감소(C3/C4 56/2 mg/dL, 정상치: 86-160/17-45 mg/dL), 루푸스 항응고인자(lupus anticoagulant) 양성, 항핵항제(FANA) 양성(1:320 speckled pattern)을 보였고 기타 검사 소견으로는 항SS-A항체, 항SS-B항체, 항ENA (RNP)항체 음성, ANCA 음성 소견을 보였다. 직접 쿰스 검사와 간접 쿰스 검사 모두 음성이었다. 뇌척수액 검사상 WBC 11/mm3 (정상치: < 5), 배양검사 음성으로 비감염성 수막염 소견이었다.
방사선 소견: 흉부 단순촬영에서 양측성 흉수가 관찰되었다. 외부 병원의 조영증강 복부 전산화 단층촬영에서 양측성 신우신염과 비장 경색 의증 또는 외상에 의한 비장의 열상 의증 소견을 보였다(Fig. 1). 신경학적 이상에 대한 뇌 자기공명영상에서는 특이소견은 없었다.

Abdominal computed tomography (CT) scan shows a diffuse, enlarged spleen with hypodense lesions and perisplenic fluid collection suggestive of a splenic laceration (A), and multifocal wedge-shaped hypodensity lesions (arrows) in both kidneys suggestive of acute pyelonephritis (B).
골수 천자 및 생검 소견: 추적 혈액 검사에서 악화된 범혈구 감소증 소견을 보여 골수 검사를 시행하였다. 골수 흡입 검사상 적혈구계 세포와 거대핵세포의 수는 적절하였으며, 골수계 세포의 수와 성숙도도 적절하였다. 하지만 조직구의 증가와 함께 혈구탐식의 양상이 관찰되었다.
치료 및 경과: 이상의 검사 결과를 바탕으로 1997년에 미국류마티스학회(American College of Rheumatology, ACR)에서 제시된 기준에 따라 구강궤양, 혈액학적 이상(백혈구감소증, 혈소판감소증), 루푸스 항응고인자(lupus anticoagulant) 양성, 항핵항제(FANA) 양성(1:320 speckled pattern)을 만족하여 SLE로 진단하였다. 또한 발열, 비장종대, 페리틴증가, 중성지방 증가, 골수검사 상의 혈구탐식소견, 추적 혈액 검사에서 악화된 범혈구 감소증 등으로 hemophagocytic lymphohistiocytosis-2004 (HLH-2004)의 진단기준에 만족하여 SLE에 동반된 2차성 혈구포식성 림프조직구증(Hemophagocytic lymphohistiocytosis, HLH)으로 진단하였다. 환자는 prednisolone 50 mg qd와 hydroxychloroqhine 200 mg qd로 치료를 시작하였으나 혈소판감소증이 15,700/mm3까지 악화되고 피부의 점상 출혈이 동반되어 면역글로불린(IVIG)을 400 mg/kg/day의 용량으로 5일간 추가 투약하였다. 하지만 이후에도 만족스러운 치료 효과를 보이지 않아서 methylprednisolone pulse therapy (1,000 mg/day) 3일간 시행하였고 이후 발열 및 범혈구감소증의 호전을 보였으며 기타 임상경과의 호전을 보였다. 복부 전산화 단층촬영상 보인 비장의 병변에 대해서는 비장경색보다는 외상에 의한 비장의 열상으로 판단하고 경과 관찰 및 복부 전산화 단층촬영 추적을 계획하였다. 환자는 prednisolone을 점차 감량하였고, 최종적으로 prednisolone 25 mg qd, hydroxychloroqhine 200 mg qd를 유지하며 퇴원하였다. 이후 외래에서 cyclosporin을 추가했고, 이후 추적관찰하던 중 퇴원 후 약 3주 뒤 호흡곤란, 얼굴 부종, 단순 흉부 촬영상 흉수의 증가를 보여 재입원을 하였다. 재입원 후 prednisolone, cyclosporin을 증량하였고, azathioprine을 추가하였지만 호흡곤란이 악화되고 복부의 불편감을 호소하여 복부 전산화 단층촬영을 시행하였다. 복부 전산화 단층촬영 결과 비후된 비장의 파열을 동반한 미만성 침윤성 병변이 관찰되어 기저질환으로 림프종을 의심하였다(Fig. 2). 이에 이전 입원시 시행하였던 골수검사 검체에 추가적인 면역조직화학 염색을 시행하였고 결과상 혈관 내의 비정형 세포에서 CD20에 양성소견을 보였고, Factor-VIII R Ag에 대해서는 혈관에 양성소견을 보였다(Fig. 3). 이러한 소견을 바탕으로 혈관내B대세포림프종(IVLBCL)으로 진단하고 추가적으로 양전자방출단층촬영술(positron emission tomography, PET)을 시행하였으며 우측 폐하엽과, 비장, 골수의 림프종의 침윤으로 보이는 병변을 확인하였다(Fig. 4). 이후 환자는 IVLBCL의 치료를 위해 혈액종양내과로 전과되었고, rituximab, cyclophosphamide, adriamycin, vincristine, prednisolone (R-CHOP) 항암화학치료를 2회 실시하였다. 하지만 환자는 두 번째 항암화학 치료 후 부작용으로 골수억제 및 호중구 감소성 발열소견을 보였으며 동반된 폐렴 및 패혈증으로 사망하였다.

Follow-up axial (A) and coronal (B) abdominal CT scans show splenomegaly with diffuse infiltrative hypodense lesions and lateral perisplenic ruptures, suggesting hemophagocytic syndrome, and ruling out an underlying lymphoma. CT, computed tomography.
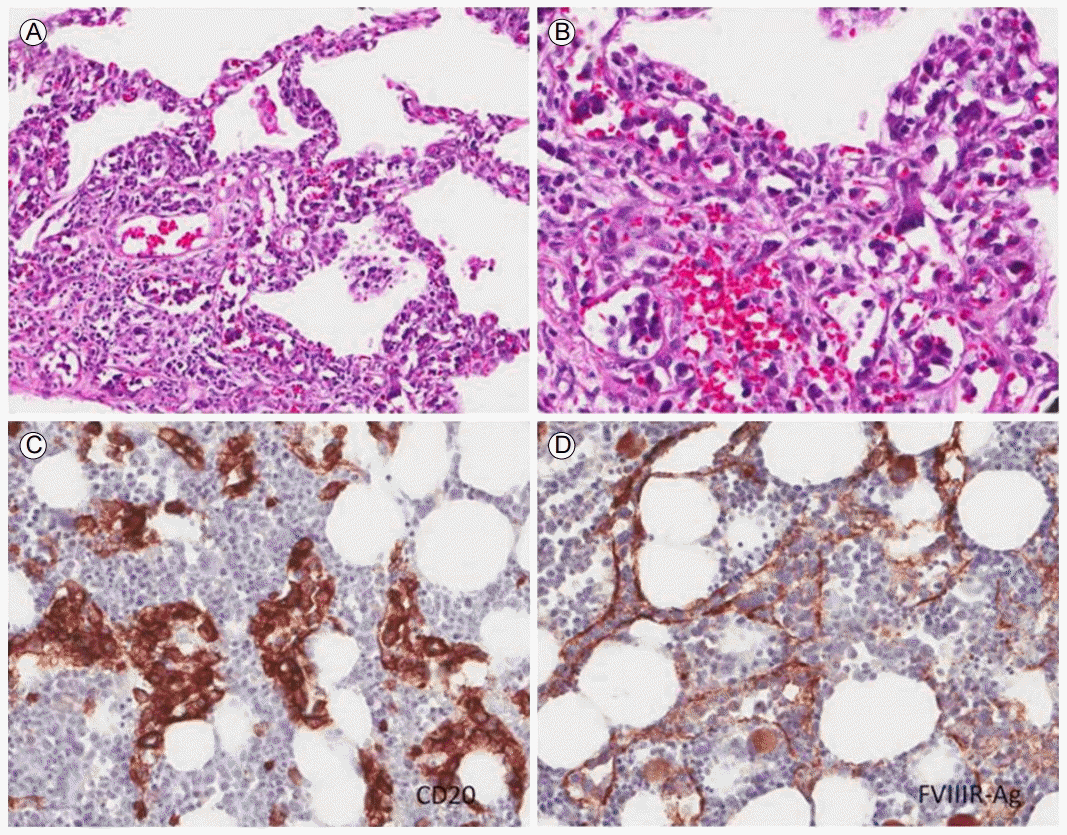
Histopathology of the bone marrow. (A) Bone marrow was hypercellular with scattered atypical cells in clusters (hemotoxylin and eosin [H&E] stain, ×200). (B) Medium to large lymphoid tumor cells with vesicular chromatin infiltrated the intrasinusoidal space (H&E stain, ×400). (C) The intravascular lymphoma expressed CD 20 (immunohistochemical staining, ×400). (D) Factor VIII stain indicating the intravascular location of the tumor cells (×400).
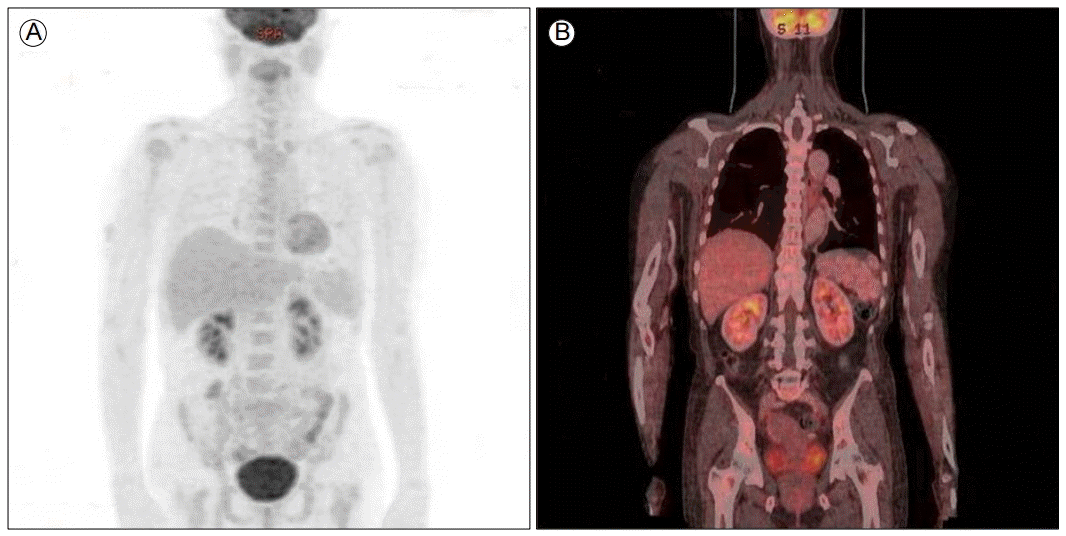
The scout film (A) and coronal plane (B) positron emission tomography-computed tomography (PET-CT) scans show multifocal low-attenuated lesions in the spleen and diffuse increased glucose metabolism in the bone marrow.
고 찰
SLE는 전신을 침범하는 자가면역질환으로 다양한 경과 및 임상소견을 보이므로 진단시 다른 질환과의 면밀한 감별진단이 요구된다. 이전부터 SLE 진단기준으로 가장 많이 이용된 것은 1982년 미국류마티스학회(American College of Rheumatology, ACR)에서 제시된 분류기준으로, 이것은 15년 동안 표준으로 사용되었고 1997년 한 차례 개정이 된 이후에도 지속적으로 사용되었다[4]. 본 증례에서도 내원시 1997년 ACR의 분류기준에 근거하여 SLE로 의심을 하게 되었는데, 이는 임상 연구 수행을 위해서는 양호하나 진단기준으로 사용하기에는 다음과 같은 한계점들 보여왔다. 첫째, 총 11개의 항목 중 광과민성, 뺨 발진, 원판상 발진, 구강궤양 등 4가지가 피부점막증상으로 편향된 점, 둘째, 세 가지 이하 기준을 만족하는 비전형적 초기 환자 진단의 어려움, 셋째, 신경학적 이상 항목에서 발작과 정신병 두 가지만 반영된 점, 넷째, 질병 활성도를 반영하는 보체 검사의 부재, 마지막으로 11개 항목에 가중치가 없이 똑같이 취급되었다는 점이다[5]. 이처럼 기존의 1997년 ACR의 분류기준은 장기 침범이 명확하지 않은 SLE 초기 환자를 진단하는데 한계점이 있으며, 한편으로는 SLE의 임상양상이 명확하지 않아도 분류기준을 만족하여 다른 질환을 SLE로 오인하게 되는 경우가 종종 발생하였다. 특히 진단시 매우 중요하게 사용되는 자가항제의 경우 대부분의 SLE 환자의 혈청에서 검출되지만 이는 다른 면역질환 및 악성종양에서도 나타나 특이도 면에 부족한 점이 있으며, 한편으로는 질환 특이 항체로 알려진 항dsDNA항체, 항Sm항체 등은 모든 SLE 환자에서 나타나는 것은 아니므로 민감도 면에서도 역시 부족한 면이 있었다[2]. 본 증례의 환자가 최종 진단된 IVLBCL는 NHL의 드문 아형으로, 1992년 Swissa 등에 의해 보고된 바에 따르면 NHL로 치료받은 환자들의 항핵항체 양성률은 최대 32%였으며, 2003년 Guyomard 등[6]은 NHL 환자를 대상으로 림프종 치료 시작 전 측정한 항핵항체 양성률을 19%로 보고하였고 항핵항체 양성의 환자 중 약 28%에서 기존의 자가면역 질환에서도 자주 나타나는 관절증상, 피부증상 등의 임상증상을 보였다고 보고하였다. 또한 본 증례의 환자에서 관찰된 혈구포식성 림프조직구증(HLH)은 발열, 범혈구 감소증, 페리틴의 증가 및 골수나 간, 림프절 등에서의 혈구 탐식을 특징으로 하는 드문 질환으로 약물, 바이러스 감염 등에 의해 이차적으로 발생할 수 있고, 림프종 등의 악성종양과 SLE 등의 여러 자가면역질환에서도 공통적으로 발견되기 때문에 HLH 진단시 기저질환에 대한 감별이 필요하다[7]. 이와 같은 1997년 ACR의 분류기준에서 보였던 문제점들을 보완하기 위해 2012년에 SLE 전문가 그룹으로 구성된 Systemic Lupus Erythematosus International Collaborating Clinics (SLICC)에서 새로운 SLE 분류기준을 제시하였다. 새로운 기준의 특징은 첫째, 신생검으로 확진된 루푸스 신염에 가중치를 부여하였고, 둘째, SLE 활성도의 척도 중 하나인 저보체혈증의 포함 및 항인지질항체 검사로 항beta2GP1항체가 추가되었으며, 셋째, 신경학적 양상에 경련과 정신병 외에 다발성단신경염, 척수염, 말초 혹은 두개 신경병증으로 영역을 확장한 점이라 할 수 있다. 2012년 SLICC의 분류기준을 검증한 결과 1997년 ACR의 분류기준과 비교시 분류오류는 현저히 줄일 수 있었고, 민감도는 개선되고(94% vs. 86%), 특이도는 거의 동등한 소견(92% vs. 93%)을 보였다[2,8]. 하지만 본 증례의 경우에는 2012년에 SLICC에서 제시된 기준에 맞춰 보아도 구강궤양, 신경학적증상(비감염성 수막염, 중추신경마비), 백혈구감소증, 혈소판감소증, 항핵항체 양성, 보체감소 등을 만족하여 SLE로 진단을 할 수 있기 때문에 새로운 분류기준이 특정 장기의 침범이 저명하지 않고 비전형적인 증상을 주로 보이는 환자의 경우에 있어서는 악성종양 및 다른 자가면역질환과의 감별진단의 모든 어려움을 해결한 것은 아니라고 할 수 있다. 한편, IVLBCL은 NHL의 한 종류로 혈관내 악성 림프구의 증식을 특징으로 하는데, 다른 림프종과 달리 발병 초기에 림프절 병변을 거의 동반하지 않으며 골수, 비장, 간, 말초혈액 등의 침범이 드문 반면 주로 중추신경계와 피부를 침범하는 것으로 알려져 있다. 중추신경계 증상으로 두통, 경련, 감각이상 등을 보일 수 있고 피부증상으로 홍반성 결절, 하지의 궤양성 병변 등을 보일 수 있으나 이 또한 비 특이적이거나 자가면역질환에서도 관찰되는 증상들이며 심지어 중추신경계와 피부 증상보다 발열, 체중감소, 전신쇠약 등과 같은 비 특이적인 전신증상만을 보이는 경우가 있어 감염성 질환, 다른 악성종양, 자가면역질환 등으로 오인되는 경우가 종종 있다[1,9]. IVLBCL의 치료는 중등도 또는 고도의 림프종에 준하여 주로 CHOP 등의 항암화학요법을 실시하며 CD20을 표적으로 하는 rituximab의 병용요법이 도입되면서 이전보다 치료 반응률이 상승되고 생존기간의 연장 면에서 개선을 보였지만, 앞서 언급한 진단적 어려움과 빠르고 공격적인 질병경과로 인해 여전히 좋지 않은 예후를 보인다[10].
본 증례의 환자는 내원 당시의 1997년 ACR의 기준과 이후의 2012년 SLICC의 기준을 모두 만족하여 SLE로 진단되었지만, 이후 스테로이드와 면역억제제의 일반적인 SLE의 치료에도 불구하고 만족할 만한 임상경과의 호전을 보이지 않았다. 이후 골수검사의 추가적인 면역조직화학염색을 통해 IVLBCL로 진단 후 R-CHOP 항암화학 치료를 시행하였지만 합병증이 발생하여 발병 후 약 4개월 만에 사망하였다. 따라서 본 증례와 같이 특정 장기에 대한 증상이 명확하지 않고 비특이적인 전신증상을 보이는 환자에서는 자가면역질환의 진단기준에 부합하더라도 적절한 치료 반응을 보이지 않고 악화되는 경우에는 악성종양 등과의 면밀한 감별진단이 중요하다고 할 수 있다.