특발성혈소판감소성자반증 환자에게 발생한 폐혈전색전증 1예
A Case of Pulmonary Thromboembolism in a Patient with Idiopathic Thrombocytopenic Purpura
Article information
Abstract
일반적으로 특발성혈소판감소성자반증과 같이 혈소판감소증이 있는 경우 출혈합병증이 아닌 심부정맥혈전증이나 폐혈전색전증과 같은 혈전성 질환이 발생하는 것은 드문 현상이다. 하지만 본 증례와 같이 특발성혈소판감소성자반증 환자에서 혈전증이 합병되면서 항인지질증후군이 병발하는 경우가 있으므로 의심증상이 있을 때에는 적극적인 진단 및 치료가 필요하다. 저자들은 특발성혈소판감소성자반증 환자가 하지혈전증 및 폐혈전색전증이 발생하면서 항인지질증후군이 진단되고, 항응고제 및 면억억제제를 동시에 투여하면서 효과적으로 치료된 증례를 경험하였기에 문헌고찰과 함께 보고하는 바이다.
Trans Abstract
Idiopathic thrombocytopenic purpura (ITP) is an autoimmune blood disorder characterized by thrombocytopenia. Common clinical manifestations include bleeding events. Rarely, thrombotic complications may develop in ITP. A 43-year-old man was admitted with dyspnea. His platelet count at admission was 48,000/mm3. The patient had a history of ITP diagnosed 12 years earlier and had been treated with low-dose steroids. Two months before admission, he had been diagnosed with deep vein thrombosis and treated only with clopidogrel due to severe thrombocytopenia. Chest computed tomography showed filling defects in both pulmonary arteries. In the workup for precipitating factors, only lupus anticoagulant was positive. The concomitant administration of warfarin and methylprednisolone was used to treat the pulmonary thromboembolism and ITP, respectively. Six months later, the lupus anticoagulant test remained positive. The patient was confirmed to have a pulmonary thromboembolism due to antiphospholipid syndrome, which might be related to the underlying ITP. After 10 months, his symptoms and radiological findings had improved. (Korean J Med 2011;81:251-256)
서 론
폐혈전색전증(pulmonary thromboembolism)은 주로 심부 정맥 혈전증(deep vein thrombosis)에 이차적으로 발생한다. 따라서 폐혈전색전증은 발생 기전이 정맥혈전증과 유사하여 혈류정체(vein stasis), 혈관 벽 손상(endotheilal injury) 및 과응고 상태(hypercoagulability) 등에 의해 유발된다[1]. 2003년 국내에서 시행한 폐혈전색전증의 전국 실태 조사에서[2] 혈액응고병증(coagulopathy)은 전체 발생 원인의 7.3%를 차지하였다. 특히 본태성혈소판증가증이나 진성적혈구증과 같은 골수증식성 질환은 각각 0.5% 빈도로 매우 드물었다. 이러한 골수증식성 질환에서 혈전증이 호발하는 이유로는 과혈구증으로 인해 혈액의 점도가 상승하여 혈류가 정체되고, 혈소판이 비정상적으로 활성화되는 점 등이 알려져 있다 [3,4].
따라서 혈소판이 감소되어 있는 상황에서 혈전증이 발생하는 것은 그다지 흔하지 않다. 특발성혈소판감소성자반증은 혈소판 감소를 특징으로 하며 출혈성 임상 증상을 보이는 자가면역질환이다[5]. 저자들은 특발성혈소판감소성자반증으로 혈소판이 저하된 환자에게 하지정맥혈전증 및 폐색전증이 발생된 드문 경험을 하였기에 증례 보고와 함께 문헌고찰을 한다.
증 례
환 자: 이○동, 43세 남자
주 소: 5시간 전 시작된 흉부 불편감 및 호흡곤란
현병력: 환자는 12년 전 혈액내과에서 특발성혈소판감소성자반증을 진단받고 methylprednisolone 4 mg 유지하였다. 2개월 전 갑자기 발생한 우측하지의 파행으로 시행한 하지 도플러 초음파에서 하지정맥혈전증이 발견되어 외래에서 clopidogrel 75 mg 및 methylprednisolone 12 mg 복용하던 중, 입원 당일 발생된 흉부 불편감과 호흡곤란으로 입원하였다.
과거력 및 사회력, 가족력: 흡연력은 10갑년으로 현재 흡연 중이며, 다른 과거력 및 가족력상 특이소견 보이지 않았다.
이학적 소견: 환자의 전신상태 및 영양 상태는 양호하였다. 혈압은 120/70 mmHg, 맥박 95회/분, 호흡수 20회/분이었으며, 체온은 36.5℃이었다. 환자의 피부 및 두부 이학적 검사에서 이상이 없었고, 경부 림프절은 촉지되지 않았다. 흉부 청진시 양측 폐야의 호흡음은 정상이었고, 심음은 규칙적이었으며 잡음은 없었다. 양하지에 경도의 부종이외의 특이 소견은 없었다.
검사실 소견: 말초혈액검사에서 백혈구 19,950/mm3 (호중구 77.9%), 혈색소 15.6 g/dL, 헤마토크리트 48.2%, 혈소판은 48,000/mm3이었다. 생화학 검사상 총 단백 7.4 g/dL, 알부민 4.2 g/dL, 아스파라진산아마이드효소(AST) 17 IU/L, 알라닌아미노전이효소(ALT) 21 IU/L, 공복 혈당 95 mg/dL, 혈중요소질소 15.9 mg/dL, 크레아틴 1.04 mg/dlL, 총 빌리루빈 0.73 mg/dL이었다. 적혈구 침강속도는 6 mm/h, CRP 18.57 mg/L이었고, 대기 중 동맥혈 가스 검사는 pH 7.386, PaCO2 35.6 mmHg, PaO2 82.7 mmHg, HCO3- 20.9 mmHg, 산소포화도 97.7%였다. 프로트롬빈시간(prothrombin time) 12.0초, 활성화부분트롬보플라스틴시간(aPTT) 25.8초로 모두 정상 범위였다. D-dimer는 345 μg/L (참고치 0-200 μg/L)로 상승되어 있었다. VDRL은 음성 및 류마토이드 인자는 정상범위였고, 항핵항체(antinuclear antibody)는 음성, C3 및 C4 농도는 정상이었다. 항혈소판 항체는 음성이었다. 섬유소원 350 mg/dL (참고치 196-391 mg/dL), antithrombin III 83.6% (참고치 8.1-119.8%)이었고, C 단백질 및 S 단백질 항원은 각각 94% (참고치 70-130%), 70% (참고치 65-140%)이었다. 항카디오리핀 항체와 베타-2 당단백 I 항체는 음성이었으나, 루푸스 항응고 인자는 양성이었다.
방사선 검사 소견: 내원 2개월 전 시행한 하지도플러초음파에서 우측 대퇴 정맥에서 슬와 정맥(popliteal vein)까지 급성 심부정맥혈전증 소견이 관찰되었다(Fig. 1). 입원 시 촬영한 단순흉부사진에서는 특이소견은 없었으나, 흉부 전산화 단층 촬영에서 우중엽, 우하엽 및 좌하엽으로 가는 폐동맥에 음영 결손의 혈전들이 확인되었다(Fig. 2). 추적관찰한 하지도플러초음파에서는 이전 병변 주변으로 측부 혈관(collateral vessels)이 생성되어 정맥혈전이 만성화된 양상을 보였다.
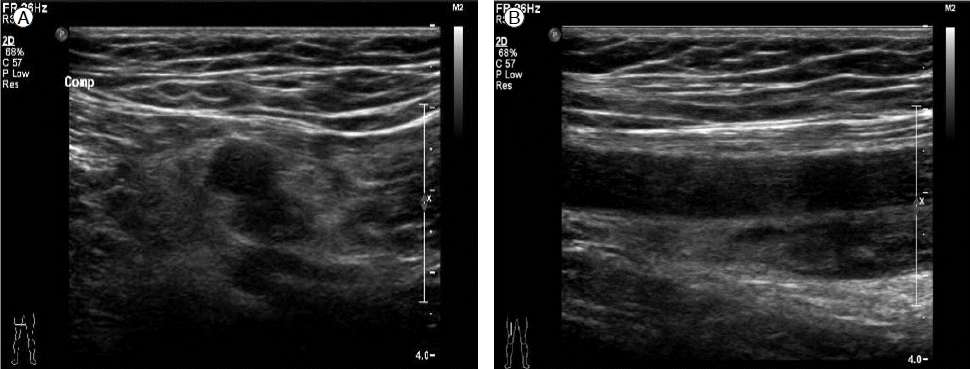
Doppler ultrasonography of both extremities two months before admission shows deep vein thrombosis involving the right femoral and popliteal arteries.
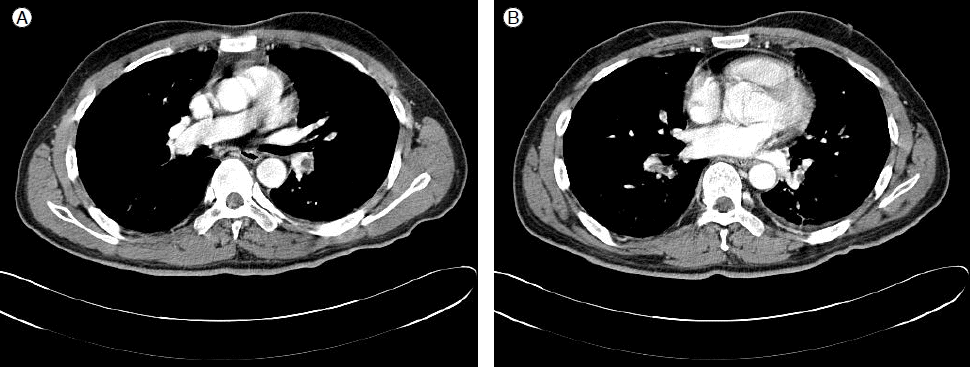
Chest computed tomography at admission shows filling defects in the branch of the right interlobar artery to the right middle/lower lobe and interlobar/segmental branch of the left inferior pulmonary artery.
임상경과: 폐혈전색전증이 진단되어 저분자량헤파린을 1 mg/kg으로 하루 2회 피하주사하였고, 3병일째부터 증상은 호전되어 경구 와파린(warfarin) 5 mg을 복용하면서 퇴원하였다. 퇴원 6개월 후 시행한 루푸스항응고인자가 지속적으로 양성 소견을 보여 항인지질증후군으로 최종 진단하였다. 현재 환자는 와파린 4 mg과 스테로이드(methylprednisolne) 12 mg로 치료 중이며, 10개월 후 시행한 흉부 컴퓨터 단층촬영에서 폐혈전색전증은 모두 소실되었다(Fig. 3).

Chest computed tomography 10 months later shows complete resolution of the pulmonary thromboembolism in both lungs.
고 찰
특발성혈소판감소성자반증은 면역 기전의 이상으로 혈소판 감소증이 유발되는 자가 면역 질환으로, 발생 빈도는 미국에서는 연간 100만명 당 50-100명 정도로 알려져 있다[5]. 무증상부터 자반증, 코피, 뇌출혈에 이르기까지 다양한 정도의 출혈 증상을 보이고, 진단은 혈소판 감소증을 일으킬 수 있는 약물, 감염 및 혈액 질환 등을 배제함으로써 이루어진다. 출혈 양상이 대표적인 특징인 특발성혈소판감소성자반증에서 혈전성 합병증이 동반되는 경우는 흔하지 않으며, 국내 조사에 따르면 뇌경색증, 관상동맥색전증 등이 병발한 증례[6,7]는 있으나 본 예와 같이 심부 정맥 혈전증이나 폐혈전색전증이 보고된 적은 없다.
특발성혈소판감소성자반증이 중증도 이상의 혈소판 감소증에도 불구하고 이와 같은 혈전증이 발생하는 기전에 대해 명확하게 정립되지는 않았지만, 현재까지 나온 가설들을 살펴보면 다음과 같다. 첫째, 특발성혈소판감소성자반증의 치료와 관련된 경우이다. 스테로이드, 면역글로불린, 비장절제술 등의 치료로 낮았던 혈소판 수치가 갑자기 증가하면 혈장의 점도가 상승하여 혈전이 생길 수 있다는 것이다[8]. 둘째, 특발성혈소판감소성자반증의 20-60%에서 양성으로 보고되는 혈소판에 대한 자가항체와 관련된 가설이다. Fruchter 등[9]은 특발성혈소판감소성자반증 환자에서 치료와 무관하게 심근경색이 발생한 증례를 보고하면서, 혈소판과 혈관 내피세포 사이에 항원 유사성(antigen mimicry)이 있어 혈소판에 대한 자가항체가 혈관 내피세포의 항원에도 작용하여 혈전을 유발하여 심근경색을 유발했을 것으로 추정하였다. 마지막으로, 루푸스 항응고 인자(lupus anticoagulant), 항카디오리핀 항체(anticardiolipin antibody), 항베타2-당단백 I (anti β2- glycoprotein I) 등과 같은 항인지질 항체가 관여한다는 가설이다. 보고에 따르면, 특발성혈소판감소성자반증 진단 시 26-38%의 빈도로 루푸스 항응고 인자나 항카디오리핀 항체가 양성으로 나타났다[10,11].
본 증례에서 환자는 폐혈전색전증 진단 당시 스테로이드를 유지 용량으로 사용하고 있었지만 혈소판수가 48,000/mm3으로 입원 수개월 전과 비교하여 큰 차이를 보이지 않아 치료와 연관되어 발생한 것으로 생각되지는 않는다. 왜나하면 스테로이드를 사용하는 ITP 환자들은 대부분 스테로이드 용량을 변경한 뒤 혈소판이 증가할 때에 혈전증이 발생하기 때문이다. 또한 항혈소판 항체는 음성이었으므로 위의 두 번째 가설인 항체 관련 혈관내피손상의 기전으로 설명하기는 어렵다. 한편, 환자는 12년 전 특발성혈소판감소성자반증을 처음 진단 당시의 검사 결과는 확인할 수 없으나, 금번 폐혈전색전증 발병 시 시행한 검사에서 루푸스 항응고 인자가 양성이었으므로 세 번째 가설과 가장 합당한 것으로 생각된다. 이후 환자는 6개월 뒤 재시행한 루푸스 항응고 인자도 양성 소견을 보였다. Sapporo 진단 기준[12]에 따르면, 임상적으로 혈전성 질환이 있으면서 검사실 검사에서 루푸스 항응고 인자나 항카디오리핀 항체가 6주 이상의 간격으로 두 번 이상 측정시 지속적으로 양성이 나오는 경우 항인지질증후군으로 진단할 수 있어 본 환자 역시 항인지질증후군이 발생하였음을 확인하였다. 보고마다 다소 차이가 있으나, Diz-Küçükkaya 등[11]는 특발성혈소판감소성자반증 환자 82명의 항인지질 항체를 조사하고 5년간 관찰한 결과 14명의 환자에서 항인지질증후군이 발생하였다. Kravitz 등[13]은 특발성혈소판감소성자반증, 혈전성혈소판감소성자반증, 헤파린유발성혈소판저하증 및 항인지질증후군 등 네 가지 질환 모두, 자가항체에 의한 면역 기전에 의해 혈소판 감소증이 유발되기 때문에 동일한 환자에서도 위의 두 질환이 병발 혹은 한 질환이 다른 질환으로 진행될 수 있음을 자가면역의 만화경 (kaleidoscope of autoimmunity)의 개념으로 설명하였다. 자가면역은 유전적, 면역학적, 호르몬적, 환경적 요소 등이 복합적으로 관여하게 되며, 이를 자가면역의 모자이크즘(mosaic of autoimmunity)이라고 하며[14], 한 자가면역질환에서 위와 같이 영향을 미치는 요소의 변화를 통해 다른 자가면역질환이 발병하는 현상을 kaleidoscope 현상이라고 한다[15]. 실제로 특발성혈소판감소성자반증 환자에서 비장 절제술 이후 쇼그렌 증후군과 원발성담즙성간경변이 발생한 사례[16], 면역혈소판감소증 환자에서 비장 절제술 이후 자가면역성간염이 발생되었던 예[17] 등이 보고되었다. 따라서 본 증례는 특발성혈소판감소성자반증으로 진단 및 치료를 받고 12년 뒤 심부 정맥 혈전증 및 폐혈전색전증이 발생하면서 항인지질증후군이 병발되었음을 확인한 경우이다. 한편, 특발성혈소판감소성자반증 환자에서 추적관찰 중 항인지질증후군이 발생한 경우 초기 진단 당시 루푸스 항응고 인자가 양성이었고 이후 지속적으로 양성인 경우가 많아 추후 항인지질증후군이 병발할 가능성을 예측하는 인자로 루푸스 항응고 인자를 제시하는 연구들이 있다[10,11,18]. 아직까지는 특발성혈소판감소성자반증을 진단할 때 항인지질 항체 검사가 필수적이지는 않으나 본 증례와 같이 폐혈전색전증 및 항인지질 증후군 발생 및 이로 인한 치명률을 고려할 때 초기 진단 시 선택적으로 검사를 시행해 볼 수 있을 것이며, 향후 이와 같은 항인지질 항체의 임상적 의미에 대한 대규모 전향적 연구가 필요하겠다.
이와 같이 특발성혈소판감소성자반증으로 혈소판 저하증이 있는 상황에서 혈전증을 치료하는 데는 출혈 위험성이 있어 쉽지 않다. 본 증례에서도 초기에 외래에서 심부 정맥 혈전증이 진단되었을 당시 치료로 항혈소판제제인 clopidogrel을 사용한 것도 같은 이유 때문이었다. 그러나 치료에도 불구하고 폐혈관색전증까지 발생하여 항응고제를 시작하게 되었고, 특별한 출혈 합병증 없이 효과적으로 치료되었다. Iwahara 등[19]은 루푸스 항응고 인자를 가지는 특발성혈소판감소성자반증 환자에서 면역억제치료(immunosuppressive therapy)와 항혈전 치료(antithrombotic therapy)를 병행하여 효과적으로 치료한 증례를 보고하였다. 본 환자 역시 항응고치료로 경구 와파린 3 mg와 함께 면역억제제인 methylpredisolone 4 mg을 함께 복용하면서 외래 추적관찰 10개월 째 혈소판 수, 임상 증상 및 방사선학적인 소견 모두 안정화된 상태이다.