


INTRODUCTION
Pulmonary placental transmogrification (PT) is a rare benign lesion first described by McChesney in 1979 [1]. PT is defined as placental villi-like papillary structures in the lung parenchyma; however, this tissue contains no biological components of the placenta despite the morphological similarity [2]. It is composed of epithelial cells, proliferating vessels, inflammatory cells, and fat. Disease pathology is characterized by the formation of papillary structures similar to placental villi surrounding the pulmonary epithelium [3,4]. Radiologically, PT of the lung shows mainly bullous changes [4]. Although many hypotheses have been proposed to describe the pathogenesis of PT, the evidence remains unclear [3,5]. We herein report a case of PT in a 66-year-old woman who presented with a single nodule.
CASE REPORT
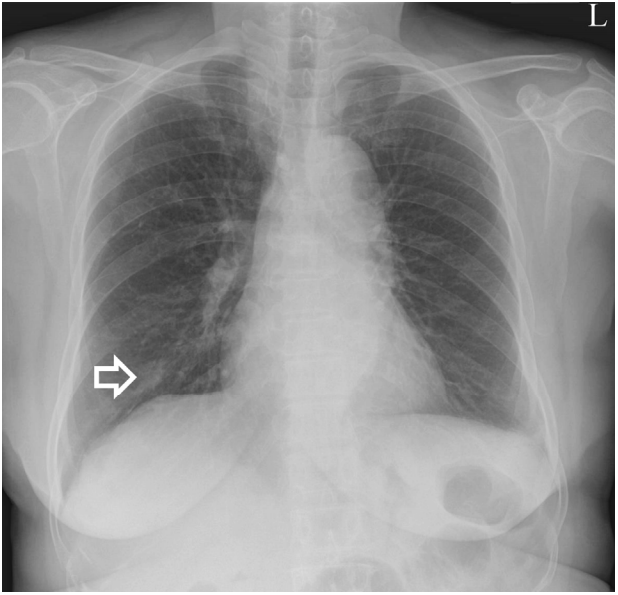
A 66-year-old woman presented with a small nodular lesion on chest radiography performed during a routine health examination. She did not complain of respiratory or systemic symptoms. She was a nonsmoker with well-controlled diabetes and hypertension over a 10-year period. There was no other notable familial or occupational history. Physical examination and laboratory tests were unremarkable, and no acid-fast bacilli were detected on sputum culture. Chest radiography showed a focal ill-defined nodular opacity in the right lower lobe zone (Fig. 1). Chest computed tomography (CT) revealed a 17-mm lobulated and focal irregular mass with fissural retraction in the right lower lobe anterobasal segment of the lung, suggestive of lung cancer such as adenocarcinoma in situ (Fig. 2A and B), and two other lung nodules (<5 mm) in the right upper lobe anterior segment and right lower lobe superior segment (Fig. 2C and D). There were no contralateral nodules, enlarged lymph nodes, or effusions. Pulmonary function testing showed a forced expiratory volume during 1 second (FEV1) of 1.89 L (90% of the predicted value), a forced vital capacity (FVC) of 2.69 L (96% of the predicted value), and a diffusion capacity of carbon monoxide of 20.6 mL/mm Hg/min (132% of the predicted value). The FEV1/FVC ratio was 0.70.
A CT-guided percutaneous needle aspiration biopsy of the right lung nodule was performed for histological confirmation. The two smaller nodules could not be characterized. Because the biopsy was not performed due to their small size and location; regular follow-up CT was planned to monitor these lesions. Microscopy revealed a papillary structure resembling chorionic villi on scanner view (Fig. 3A). The papillary projection was lined by cytotrophoblast- like cells with a single nucleus and basophilic cytoplasm and syncytiotrophoblasts with multiple small nuclei and eosinophilic cytoplasm. An edematous stroma with bland ovoid interstitial cells was evident (Fig. 3B). The cells of the papillary structure were strongly positive for ╬▓-hCG (Fig. 3C), but negative for TTF-1 (Fig. 3D). These characteristic histological and immunohistochemical findings led to a diagnosis of PT of the lung. The patient did not complain of symptoms, and there was no interval difference on chest radiography and chest CT during the 1-year follow-up after the diagnosis.
DISCUSSION
PT of the lung is a rare disease that has been adequately documented in only 30 cases [4]. Clinically, this disease typically occurs in men aged 20 to 50 years and usually presents with dyspnea or pneumothorax [4,6]. We have herein presented a rare case of PT in an asymptomatic woman who presented with a single nodule. Most reported cases of PT are accompanied by emphysema and symptoms associated with pneumothorax, requiring pneumonectomy. Ferretti et al. [3] documented a case of PT presenting as a 25 mm pulmonary nodule without associated bullous emphysema. Our case is similar to that reported by Ferretti et al. [3] which involved a 17-mm nodule and no associated emphysema. Early diagnosis is important because PT requires surgical resection.
Cavazza et al. [7,8] suggested that PT may not be a variant of giant bullous emphysema, but an interstitial clear cell proliferation with secondary emphysema-like cystic change. In addition, they analyzed the immunophenotype of these clear cells and found them to be positive for CD10 and vimentin but negative for cytokeratin, actin, desmin, and S-100. In our case, interstitial clear cell proliferation was observed without emphysema-like cystic change, suggesting that PT may be associated with proliferation of lining epithelial components in the hamartomas, as suggested by Cavazza et al. [7].
Patients diagnosed with PT may remain asymptomatic for years before presenting with chest pain, dyspnea, or hemoptysis [7]. Thus, PT presents in many forms, from asymptomatic to clinically symptomatic associated with other pulmonary diseases, such as chronic obstructive pulmonary disease, repeated pneumothorax, and even respiratory distress [5,7]. Xu et al. [9] documented the frequent association of PT with pulmonary fibrochondromatous hamartomas and suggested that it may be induced by, or associated with, proliferation of lining epithelial components of the hamartomas. Therefore, this lesion may occur with non-cystic lung lesions such as fibrochondromatous hamartoma or as a solitary pulmonary nodule on routine chest images, as in our case. PT is considered benign, but a case of papillary adenocarcinoma arising in a placentoid bullous lesion has been reported [7]. Surgical resection is commonly curative and improves lung function and quality of life [7]. In our case, the patient had no respiratory complaints and there was no significant difference on follow-up chest CT. She has been advised of the possibility of changes in malignancy and is scheduled for regular outpatient follow-up visits.
In conclusion, PT is a rare pulmonary disease characterized by papillary structures similar to placental villi surrounding the pulmonary epithelium. Prior to diagnosis, most patients exhibit symptoms of severe emphysema or pneumothorax, but our patient had no symptoms. This case shows that PT may be found as an incidental solitary pulmonary nodule on routine health examination, not as an emphysema-like cystic lesion. Patients who are left untreated upon diagnosis of PT often proceed to suffer severe complications such as bullous emphysema, recurrent pneumothorax, or tension pneumothorax. Thus, early diagnosis and close follow-up is critical, as in our case.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






