


ņä£ ļĪĀ
ņāüņŚ╝ņāēņ▓┤ņÜ░ņä▒ ļŗżļéŁņŗĀ(ņØ┤ĒĢś ļŗżļéŁņŗĀ)ņØĆ Ļ░Ćņן ĒØöĒĢ£ ņ£ĀņĀäņä▒ ņŗĀņןņ¦łĒÖśņ£╝ļĪ£ ņ¢æņ¬Į ņĮ®ĒīźņŚÉ ļŗżņłśņØś ļéŁņóģņØ┤ ņāØĻĖ░Ļ│Ā ņ×Éļ×īņ£╝ļĪ£ņä£ ņŗĀĻĖ░ļŖź Ļ░Éņåī ļ░Å ņŗĀļČĆņĀäņØä ņ┤łļלĒĢśļŖö ņ¦łĒÖśņ£╝ļĪ£, ļ░£ļ│æļźĀņØĆ ļéśļØ╝ļ│äļĪ£ ņĢĮĻ░äņØś ņ░©ņØ┤Ļ░Ć ņ׳ņ£╝ļéś ļīĆĻ░£ 1,000ļ¬ģļŗ╣ 1ļ¬ģĻ╝┤ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[1]. 60ņäĖĻ╣īņ¦Ć ņĀäņ▓┤ ĒÖśņ×ÉņØś ņĢĮ ļ░śņłśĻ░Ć ņŗĀļīĆņ▓┤ņÜöļ▓ĢņØ┤ ĒĢäņÜöĒĢ£ ļ¦ÉĻĖ░ ņŗĀļČĆņĀäņŚÉ ļÅäļŗ¼ĒĢśĻ▓ī ļÉśļ®░, ĻĄŁļé┤ņŚÉņä£ Ēł¼ņäØ ĒÖśņ×ÉņØś ņøÉņØĖ ņżæ ņĢĮ 2%ļź╝ ņ░©ņ¦ĆĒĢśņŚ¼ ļŗ╣ļć©, Ļ│ĀĒśłņĢĢ, ņé¼ĻĄ¼ņ▓┤ ņŗĀņŚ╝ ļŗżņØīņ£╝ļĪ£ ĒØöĒĢ£ ņøÉņØĖņØ┤ļŗż. ļŗżļéŁņŗĀņØĆ ņ¢æņ¬Į ņĮ®ĒīźņØś ĻĄ¼ņĪ░ņĀü, ĻĖ░ļŖźņĀü Ļ▓░ĒĢ©ņØä Ļ░ĆņĀĖņś¼ ļ┐É ņĢäļŗłļØ╝ Ļ░üņóģ ņŗĀņןņÖĖ ĒĢ®ļ│æņ”ØņØä ļÅÖļ░śĒĢĀ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ļ╣äĻĄÉņĀü ņ¦łĒÖśņØś ņ┤łĻĖ░ļŗ©Ļ│äņŚÉņä£ļČĆĒä░ Ļ┤Ćļ”¼Ļ░Ć ĒĢäņÜöĒĢśļŗż.
ļŗżļéŁņŗĀņØĆ PKD1Ļ│╝ PKD2 ņ£ĀņĀäņ×ÉņØś Ļ▓░ĒĢ©ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳Ļ│Ā, ņØ┤ļōż ņ£ĀņĀäņ×ÉļŖö Ļ░üĻ░ü polycystin-1Ļ│╝ polycystin-2ļØ╝ļŖö ļŗ©ļ░▒ņ¦łņØä ņĮöļö®ĒĢ£ļŗż. ņøÉņØĖņ£ĀņĀäņ×ÉņÖĆ ņĮöļö®ļŗ©ļ░▒ņØ┤ ļ░ØĒśĆņ¦ä ņØ┤Ēøä ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņŚÉ ļö░ļźĖ ņäĖĒżļé┤ ņŗĀĒśĖņĀäļŗ¼Ļ│äņØś ļŗżņ¢æĒĢ£ ļ│ĆĒÖöņÖĆ ņØ┤ņāüņØ┤ ņĢīļĀżņ¦ĆĻ│Ā, ņ¦łļ│æņØś ņ×äņāü Ļ▓ĮĻ│╝ļź╝ ņČöņĀü ļ╣äĻĄÉĒĢĀ ņłś ņ׳ļŖö ņśüņāüļČäņäØļ▓ĢņØ┤ ĒÖĢļ”ĮļÉśļ®┤ņä£ ņ¦łļ│æņØś ņ¦äĒ¢ēņØä ļ¦ēĻĖ░ ņ£äĒĢ£ ņĢĮļ¼╝ ņÜöļ▓ĢņØ┤ ņŗ£ļÅäļÉśĻ│Ā ņ׳ļŗż. ĒŖ╣Ē׳ 2014ļģäņŚÉļŖö ņĀä ņäĖĻ│äņØś ļŗżļéŁņŗĀ ņĀäļ¼ĖĻ░ĆļōżņØ┤ ļ¬©ņŚ¼ ļŗżļéŁņŗĀ ņ¦äļŗ© ļ░Å ņ╣śļŻīņŚÉ ņ׳ņ¢┤ ļģ╝ļ×ĆņØ┤ ļÉśĻ│Ā ņ׳ļŖö ļČĆļČäņŚÉ ļīĆĒĢ┤ ĒĢ®ņØśņĀÉņØä ļÅäņČ£ĒĢśĻ│Ā Ļ░ĆņØ┤ļō£ļØ╝ņØĖņØä ņäĖņÜ░ļŖö ņØ╝ņØä ņŗ£ņ×æĒĢśņśĆļŗż[2].
ļ│Ė ņóģņäżņŚÉņä£ļŖö ņĄ£ĻĘ╝ 3ļģäĻ░ä ņ¦äĒ¢ēļÉ£ ņ×äņāüņŚ░ĻĄ¼ ļ░Å kidney disease improving global outcomes (KDIGO) controversies conference ļé┤ņÜ®ņØä ņżæņŗ¼ņ£╝ļĪ£ ļŗżļéŁņŗĀ ņ¦äļŻī ļ░Å ņ╣śļŻīņŚÉ ņ׳ņ¢┤ ņ×äņāüņĀüņ£╝ļĪ£ ņĀüņÜ® Ļ░ĆļŖźĒĢ£ ņŻ╝ņÜö ņØ┤ņŖłļź╝ ļŗżļŻ©Ļ│Āņ×É ĒĢ£ļŗż.
ņ╣śļŻīĻĄ░ņØś ņäĀņĀĢ
ļŗżļéŁņŗĀņØĆ ņ¦łļ│æņ┤łĻĖ░ņŚÉļŖö ļéŁņóģņØ┤ ņ░©ņ¦ĆĒĢśņ¦Ć ņĢŖņØĆ ņĀĢņāü ņŗĀņŗżņ¦łņØś ļ│┤ņāüņ×æņÜ®ņ£╝ļĪ£ ņŗĀņן ĻĖ░ļŖźņØ┤ ļ╣äĻĄÉņĀü ņśżļ×£ ĻĖ░Ļ░ä ņ£Āņ¦ĆļÉ£ļŗż. ņØ┤ļ¤¼ĒĢ£ ņ┤łĻĖ░ ļŗ©Ļ│äņŚÉļŖö ņŗĀņן ļ░Å ņŗĀņןņÖĖ ĒĢ®ļ│æņ”ØņŚÉ ļīĆĒĢ£ ņØ╝ļ░śņĀüņØĖ Ļ┤Ćļ”¼ņŚÉ ņ┤łņĀÉņØä ļ¦×ņČöņ¦Ćļ¦ī, ļ¦īņä▒ ņŗĀļČĆņĀäņ£╝ļĪ£ ņ¦äĒ¢ēĒĢĀ ņ£äĒŚśņØ┤ ļåÆņØĆ ļīĆņāüņ×ÉņŚÉņä£ ņŗĀĻĖ░ļŖź ļ│┤ņĪ┤ ļ░Å ļéŁņóģņä▒ņן ņ¢ĄņĀ£ļź╝ ņ£äĒĢ£ ņĢĮļ¼╝ņ╣śļŻīļź╝ ņŗ£ņ×æĒĢĀ ĒĢäņÜöĻ░Ć ņ׳ļŗż. ĒŖ╣Ē׳ ņĄ£ĻĘ╝ 10ņŚ¼ ļģä ļÅÖņĢł ļČäņ×ÉņāØļ¼╝ĒĢÖņĀüņ£╝ļĪ£ ņĢĮļ¼╝ņ╣śļŻīņØś ĒāĆĻ╣āņØ┤ ļ░ØĒśĆņ¦Ćļ®┤ņä£ ņŗĀĻĖ░ļŖź Ļ░Éņåī ņåŹļÅäļź╝ ņżäņØ┤Ļ│Ā, ļéŁņóģņØś ņä▒ņןņØä ņ¢ĄņĀ£ĒĢśĻĖ░ ņ£äĒĢ£ ņĢĮļ¼╝ņ╣śļŻī ņŚ░ĻĄ¼Ļ░Ć ĒÖ£ļ░£ĒĢ┤ņ¦ĆĻ│Ā ņ׳ņ£╝ļ®░, ņ╣śļŻīļź╝ ņŗ£ņ×æĒĢśĻĖ░ ņ£äĒĢ£ Ļ│Āņ£äĒŚśĻĄ░ ņäĀņĀĢņØ┤ ņżæņÜöĒĢśĻ▓ī ļÉśņŚłļŗż. ļŗżļéŁņŗĀņŚÉņä£ ļ¦īņä▒ņŗĀļČĆņĀäņ£╝ļĪ£ ņ¦äĒ¢ēĒĢśļŖö ņ£äĒŚśņØĖņ×ÉļĪ£ļŖö ņ┤ØņŗĀņןļČĆĒö╝(total kidney volume, TKV) [3], PKD1 ņ£ĀņĀäņ×ÉĒśĢ[4], ņĪ░ĻĖ░ņŚÉ ņ¦äļŗ©ļÉ£ Ļ▓ĮņÜ░, ņĪ░ĻĖ░ Ļ│ĀĒśłņĢĢ ļÅÖļ░śņŚ¼ļČĆ[5], ļŗ©ļ░▒ļć© ļ░Å ņ£ĪņĢłņĀü Ēśłļć©[6,7]Ļ░Ć ņ׳ļŗż.
PKD2ņŚÉ ļ╣äĒĢ┤ņä£ PKD1 ņ£ĀņĀäņ×ÉņØś ļÅīņŚ░ļ│ĆņØ┤Ļ░Ć ņ׳ņØä ļĢī ņŗĀļČĆņĀäņØä ļ╣äļĪ»ĒĢ£ ĒĢ®ļ│æņ”Ø ņ£äĒŚśņØ┤ ļåÆļŗżļŖö ņé¼ņŗżņØĆ ņØ┤ļ»Ė ņĢīļĀżņ¦ä ļ░ö ņ׳ļŗż[8]. ņĄ£ĻĘ╝ņŚÉļŖö ņŚ¼ĻĖ░ņŚÉ ļŹöĒĢ┤ņä£ PKD1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ ņżæņŚÉņä£ ņĢäļ»ĖļģĖņé░ ļ│ĆņØ┤ļ¦ī ņ׳Ļ│Ā ņżæļÅä ņĀłļŗ©ļÉśņ¦Ć ņĢŖļŖö ļ│ĆņØ┤(non-truncating mutation)Ļ░Ć PKD2ņÖĆ ņśłĒøäņØś ņ░©ņØ┤Ļ░Ć ņŚåļŗżĻ│Ā ņĢīļĀżņ¦ĆĻ│Ā[9], ņ░©ņäĖļīĆ ņŚ╝ĻĖ░ņä£ņŚ┤ļČäņäØņØś ļ░£ņĀäņ£╝ļĪ£ ņ£ĀņĀäņ×É Ļ▓Ćņé¼ņØś ļ╣äņÜ® ļīĆļ╣ä Ļ▓ĆņČ£ļźĀņØ┤ Ļ░£ņäĀļÉĀ Ļ▓āņ£╝ļĪ£ ņĀäļ¦ØĒĢśĻ│Ā ņ׳ņ¢┤ņä£, ņ£ĀņĀäņ×É Ļ▓Ćņé¼Ļ░Ć ĒÖśņ×ÉņØś ņ¦äļŗ© ļ┐É ņĢäļŗłļØ╝ ņןĻĖ░ ņśłĒøäļź╝ ņśłņĖĪĒĢśļŖö ļŹ░ ĒÖ£ņÜ®ļÉĀ Ļ░ĆļŖźņä▒ņØ┤ ļåÆļŗż. Ēśäņ×¼ ĻĄŁļé┤ņŚÉņä£ļÅä ņ£ĀņĀäņ×É Ļ▓Ćņé¼Ļ░Ć Ļ░ĆļŖźĒĢśņ¦Ćļ¦ī[10], ņĢäņ¦ü ņ×äņāüņŚÉņä£ ņśłĒøä ĒÅēĻ░Ć ļ¬®ņĀüņ£╝ļĪ£ ĒĢśĻĖ░ņŚÉļŖö ļ╣äņÜ® ļČĆļŗ┤ņØ┤ Ēü░ Ļ▓ī ĒśäņŗżņØ┤ļŗż. ļīĆņŗĀ Ļ░ĆņĪ▒ļĀźņØä ņ×ÉņäĖĒ׳ ņ▓ŁņĘ©ĒĢśļŖö Ļ▓āņØ┤ ļīĆņĢłņØ┤ ļÉĀ ņłś ņ׳ļŗż. ĒĢ£ ņŚ░ĻĄ¼ņŚÉ ļö░ļź┤ļ®┤ PKD1 Ļ░ĆĻ│äņØś Ļ▓ĮņÜ░ ļīĆļČĆļČä 55ņäĖ ņØ┤ņĀäņŚÉ ļ¦īņä▒ņŗĀļČĆņĀäņŚÉ ļÅäļŗ¼ĒĢ£ Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳ņŚłĻ│Ā, 70ņäĖ ņØ┤ĒøäņŚÉņĢ╝ Ēł¼ņäØņØä ĒĢ£ Ļ▓ĮņÜ░ PKD2 Ļ░ĆĻ│äļĪ£ ņśłņĖĪļÉśņŚłļŗż[11,12].
ļśÉĒĢ£ ņ¦łļ│æ ņ┤łĻĖ░ņŚÉ ļ╣äĻĄÉņĀü ņĢłņĀĢņĀüņ£╝ļĪ£ ņ£Āņ¦ĆļÉśļŖö ņŗĀņןĻĖ░ļŖźĻ│╝ļŖö ļŗ¼ļ”¼ ļéŁņóģņØś ņä▒ņןņŚÉ ļö░ļØ╝ ņŗĀņןņØ┤ ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņ╗żņ¦ĆĻĖ░ ļĢīļ¼ĖņŚÉ, TKVĻ░Ć ņ¦łĒÖś Ļ▓ĮĻ│╝ļź╝ ņל ļ░śņśüĒĢĀ ņłś ņ׳ļŖö ļ░öņØ┤ņśżļ¦łņ╗żļĪ£ ņØĖņĀĢļ░øĻ│Ā ņ׳ļŗż. Consortium for radiologic imaging studies of polycystic kidney disease (CRISP) ņŚ░ĻĄ¼ ļīĆņāüņ×É 241ļ¬ģņØä ņĄ£ļīĆ 8ļģäĻ░ä ņČöņĀü Ļ┤Ćņ░░ĒĢśņŚ¼ ļ¦īņä▒ņĮ®Ēīźļ│æ 3ļŗ©Ļ│äļĪ£ ņ¦äĒ¢ēĒĢśļŖö ņ£äĒŚśņØĖņ×Éļź╝ ņĪ░ņé¼ĒĢ£ Ļ▓░Ļ│╝, ņŚ░ļĀ╣, Ēśłņ▓Ł Ēü¼ļĀłņĢäĒŗ░ļŗī, ņĢīļČĆļ»╝ļć©, MCP-1 ļō▒ņØś ļŗżļźĖ ņÜöņØĖļōżļ│┤ļŗż ņ┤łĻĖ░ņŚÉ ņĖĪņĀĢĒĢ£ Ēéżļ│┤ņĀĢ ņ┤ØņŗĀņןļČĆĒö╝(height-adjusted TKV, htTKV)Ļ░Ć 600 mL/m ņØ┤ņāüņØĖ Ļ▓ĮņÜ░ ņśłņĖĪļĀźņØ┤ Ļ░Ćņן ņÜ░ņłśĒ¢łļŗż(area under curve 0.84) [13]. ļśÉĒĢ£ ņĄ£ĻĘ╝ņŚÉļŖö ļéśņØ┤ņŚÉ ļö░ļźĖ htTKV ņ”ØĻ░ĆņåŹļÅäņŚÉ ļö░ļØ╝ Ļ│Āņ£äĒŚśĻĄ░ņØä ņäĀļ│äĒĢśļŖö ĻĖ░ņżĆļÅä ņĀ£ņŗ£ļÉśņ¢┤ ĒĢ£ņĖĄ ļŗżļéŁņŗĀ ņ×äņāüņŗ£ĒŚśņŚÉ ļīĆĒĢ£ ņśüņāüņØśĒĢÖņĀü ņäĀņĀĢĻĖ░ņżĆņØ┤ ĒÖĢņŗżĒĢ┤ņ¦ĆĻ│Ā ņ׳ļŗż. CRISP ņŚ░ĻĄ¼ņ¦äņØĆ ņØ┤ļĪĀņĀüņ£╝ļĪ£ htTKV 150 mL/mļź╝ ĻĖ░ņżĆņ£╝ļĪ£ ņŚ░Ļ░ä htTKVņØś ļ│ĆĒÖöņ£©ņØä < 1.5% (1AĻĄ░), 1.5-3.0% (1BĻĄ░), 3.0-4.5% (1CĻĄ░), 4.5-6% (1DĻĄ░), > 6% (1E)ĻĄ░ņ£╝ļĪ£ ļéśļłäņ¢┤ ļČäņäØĒĢśņśĆņØä ļĢī 10ļģä ņØ┤ļé┤ņŚÉ ļ¦īņä▒ņŗĀļČĆņĀäņŚÉ ļÅäļŗ¼ĒĢśļŖö ļ╣äņ£©ņØ┤ 1AĻĄ░ 2.4%ņŚÉ ļ╣äĒĢ┤ 1EĻĄ░ņŚÉņä£ 66.9%ļĪ£ ĒśäĻ▓®Ē׳ ņ░©ņØ┤Ļ░Ć ļé©ņØä ļ░ØĒśöļŗż[14]. ĒĢ£ĒÄĖ, CRISP ņŚ░ĻĄ¼ņŚÉ ļö░ļź┤ļ®┤ ņŗĀņןĻĖ░ļŖźņØĆ TKVĻ░Ć 1,500 mLņŚÉ ļŗ¼ĒĢ£ ĒøäņŚÉņĢ╝ ņŚ░Ļ░ä ĒÅēĻĘĀ 4 mL/min ņØ┤ņāüņ£╝ļĪ£ Ļ░ÉņåīĒĢśĻĖ░ ņŗ£ņ×æĒĢ£ļŗż. ļö░ļØ╝ņä£ ņØ┤ ļ¬©ļōĀ ņ£äĒŚśņØĖņ×Éļź╝ ņĀĢļ”¼ĒĢśņŚ¼ ņĢĮļ¼╝ ņ╣śļŻīļź╝ Ļ│ĀļĀżĒĢ┤ņĢ╝ ĒĢśļŖö Ļ│Āņ£äĒŚśĻĄ░ņØä ņĀĢņØśĒĢśļ®┤ ļŗżņØīĻ│╝ Ļ░Öļŗż(Table 1).
ņ╣śļŻīĒÜ©Ļ│╝ ļ¬©ļŗłĒä░ļ¦ü
ņØ┤ņĀä ļŗżļéŁņŗĀ ņ×äņāüņŚ░ĻĄ¼ļōżņŚÉņä£ļŖö ņĢĮļ¼╝ņØś ĒÜ©Ļ│╝ļź╝ Ļ┤Ćņ░░ĒĢśņ¦Ć ļ¬╗ĒĢśļŖö Ļ▓ĮņÜ░Ļ░Ć ļ¦ÄņĢśļŖöļŹ░, ņØ┤ļŖö ņ┤łĻĖ░ ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ļŖö ņŗĀĻĖ░ļŖźņØś ļ│ĆĒÖöĻ░Ć ļ»Ėļ»ĖĒĢśļ®░, ļśÉĒĢ£ ņśłņĀäņŚÉļŖö ņśüņāüĒĢÖņĀü ļČĆĒö╝ņĖĪņĀĢļ▓ĢņØ┤ļéś Ēśłņ▓Ł ļśÉļŖö ņåīļ│Ćļé┤ ļ░öņØ┤ņśżļ¦łņ╗żĻ░Ć Ļ░£ļ░£ļÉśņ¦Ć ņĢŖņĢä ĻĘĖ ļ│ĆĒÖöļź╝ Ļ┤Ćņ░░ĒĢśĻĖ░ ņ¢┤ļĀżņøĀĻĖ░ ļĢīļ¼ĖņØ┤ļŗż. ņŗĀĻĖ░ļŖźņØś ļ│ĆĒÖöļéś ņŗĀļČĆņĀä ļÅäļŗ¼ņØ┤ļØ╝ļŖö Ļ▓░Ļ│╝ņ╣ś ļīĆņŗĀ ņ╣śļŻīĒÜ©Ļ│╝ļź╝ ļ¬©ļŗłĒä░ļ¦üĒĢĀ ņłś ņ׳ļŖö ņóŗņØĆ ļ░®ļ▓Ģņ£╝ļĪ£ ņĄ£ĻĘ╝ ņśüņāüņØśĒĢÖņĀü Ļ│äņĖĪņØä ĒåĄĒĢ£ TKVņØś ļ│ĆĒÖöņ£©ņØ┤ļéś Ēśłņ▓Ł ļśÉļŖö ņåīļ│Ćļé┤ ļ░öņØ┤ņśżļ¦łņ╗ż, ĻĘĖļ”¼Ļ│Ā ĒÖśņ×ÉĻ░Ć ņŻ╝Ļ┤ĆņĀüņ£╝ļĪ£ ļŖÉļü╝ļŖö ņéČņØś ņ¦łņØ┤ļéś ņ”ØņāüņØś ĒśĖņĀäņŚ¼ļČĆĻ░Ć ņé¼ņÜ®ļÉśĻ│Ā ņ׳ļŗż.
TKVļŖö ņ┤łņØīĒīī, computed tomography (CT) ļśÉļŖö magnetic resonance imaging (MRI)ņØä ņé¼ņÜ®ĒĢśņŚ¼ ņĖĪņĀĢĒĢĀ ņłś ņ׳ļŖöļŹ░, ņØ╝ļ░śņĀüņ£╝ļĪ£ ņ×äņāüņŗ£ĒŚśņŚÉņä£ ņ╣śļŻīņØś ĒÜ©Ļ│╝ļź╝ ļ¬©ļŗłĒä░ļ¦üĒĢśļŖö ļŹ░ļŖö ellipsoid ļ░®ļ▓Ģņ£╝ļĪ£ ņČöņĀĢĒĢ£ TKVļéś stereology ĻĖ░ļ▓ĢņØä ņØ┤ņÜ®ĒĢ£ TKVĻ░Ć ņé¼ņÜ®ļÉ£ļŗż[2]. Ellipsoid ļ░®ļ▓ĢņØĆ ņŗĀņןņØś ņןĻ▓Į, ļŗ©Ļ▓Į, Ļ╣ŖņØ┤ļź╝ ņłśņŗØņŚÉ ļäŻņ¢┤ ņČöņĀĢĒĢśļŖö ļ░®ļ▓Ģņ£╝ļĪ£ ļłäĻĄ¼ļéś ņåÉņēĮĻ▓ī ņé¼ņÜ®ĒĢĀ ņłś ņ׳ņ£╝ļ®░, CT ļśÉļŖö MRIņŚÉņä£ ņ¢╗ņØĆ ņśüņāüņŚÉņä£ ellipsoid ļ░®ļ▓Ģņ£╝ļĪ£ ņČöņĀĢĒĢ£ TKVĻ░Ć stereology ĻĖ░ļ▓ĢņØä ņØ┤ņÜ®ĒĢ£ TKVņÖĆ ļ¦żņÜ░ ņל ņØ╝ņ╣śĒĢśļ»ĆļĪ£ ņ×äņāüņ¦äļŻīņŚÉņä£ļÅä ņĀüņÜ®ĒĢśļŖö Ļ▓ī ņ¢┤ļĀĄņ¦Ć ņĢŖļŗż[14].
ņØ┤ ļ░¢ņŚÉļÅä ĻĖēņä▒ņŗĀļČĆņĀäņØ┤ļéś ļ¦īņä▒ņŗĀņ¦łĒÖśņŚÉņä£ ņŚ░ĻĄ¼ļÉśļŖö ļŗżņ¢æĒĢ£ Ēśłņ▓Ł ļśÉļŖö ņåīļ│Ć ļ░öņØ┤ņśżļ¦łņ╗żļōżņØ┤ ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ļÅä ĒÅēĻ░ĆļÉśĻ│Ā ņ׳ļŗż[15]. ĒŖ╣Ē׳ ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ņŗĀņןļéŁņóģņØ┤ ņ╗żņ¦ĆĻĖ░ ņØ┤ņĀäļČĆĒä░ Ļ│ĀĒśłņĢĢņØ┤ ĒĢ®ļ│æļÉśĻ│Ā, ņŗĀņןļé┤ ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀ ņŗ£ņŖżĒģ£ņØś ĒÖ£ņä▒ĒÖöĻ░Ć ļīĆļæÉļÉśļ®┤ņä£ ņåīļ│Ćļé┤ ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│äņØś ĒÖ£ņä▒ļÅäļź╝ ļéśĒāĆļéśļŖö ņåīļ│Ćļé┤ ļ░öņØ┤ņśżļ¦łņ╗żĻ░Ć ņŚ░ĻĄ¼ļÉśĻ│Ā ņ׳ļŗż[16]. ļśÉĒĢ£ ļéŁņóģņØś ņä▒ņןņŚÉ ļö░ļØ╝ ņŗĀņŗżņ¦łņØś ņŚ╝ņ”Ø ļ░Å ņä¼ņ£ĀĒÖöļź╝ ļ░śņśüĒĢĀ ņłś ņ׳ļŖö Ēśłņ▓Ł ļ░Å ņåīļ│Ćļé┤ ļ░öņØ┤ņśżļ¦łņ╗żņØś ņŚ░ĻĄ¼ļÅä ĒÖ£ļ░£ĒĢśļŗż[17]. Ē¢źĒøä ņśüņāüņØśĒĢÖņĀü ĒÅēĻ░ĆņÖĆ ĒĢ©Ļ╗ś ņØ┤ļ¤¼ĒĢ£ ļ░öņØ┤ņśżļ¦łņ╗żļōżņØä ņĪ░ĒĢ®ĒĢ┤ņä£ ļŗżļéŁņŗĀņØś ņ¦äĒ¢ē ņŚ¼ļČĆļź╝ ņśłņĖĪĒĢĀ ņłś ņ׳ņØä Ļ▓āņ£╝ļĪ£ ĻĖ░ļīĆĒĢ£ļŗż.
ņ╣ś ļŻī
ņ┤łĻĖ░ Ļ┤Ćļ”¼ ļŗ©Ļ│ä
ņŗĀņןĻĖ░ļŖźņØ┤ ļ╣äĻĄÉņĀü ņĀĢņāüņ£╝ļĪ£ ņ£Āņ¦ĆļÉśļŖö ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ļŖö ņŗĀĻĖ░ļŖźņØä ļ│┤ņĪ┤ĒĢśĻ│Ā ĒĢ®ļ│æņ”ØņØä ņĀüņĀłĒ׳ Ļ┤Ćļ”¼ĒĢ©ņ£╝ļĪ£ņŹ© ņŗĀĻĖ░ļŖźņØś ĻĖēņä▒ ņĢģĒÖöļź╝ ņśłļ░®ĒĢśļŖö Ļ▓āņŚÉ ņ╣śļŻīņØś ņ┤łņĀÉņØ┤ ļ¦×ņČ░ņĀĖ ņ׳ļŗż. ņĀüņĀłĒĢ£ ņłśļČä ņäŁņĘ©ļź╝ ņ£Āņ¦ĆĒĢśĻ│Ā, Ļ│ĀĒśłņĢĢņØä ņĀüĻĘ╣ņĀüņ£╝ļĪ£ ņ╣śļŻīĒĢśļ®░, ĒĢ®ļ│æņ”ØņØä ņĪ░ĻĖ░ņŚÉ ņ¦äļŗ©ĒĢśĻ│Ā ņ╣śļŻīĒĢśļŖö Ļ▓āņŚÉ ļīĆĒĢ┤ ņĢīņĢäļ│┤Ļ▓Āļŗż.
ņĀüņĀłĒĢ£ ņłśļČä ņäŁņĘ©
ļŗżļéŁņŗĀņŚÉņä£ļŖö ĒĢŁņØ┤ļć© ĒśĖļź┤ļ¬¼ņØĖ ļ░öņåīĒöäļĀłņŗĀ(arginine vasopressin, AVP)ņØ┤ ņ”ØĻ░ĆļÉśņ¢┤ ņ׳ņ£╝ļ®░, ņØ┤ļŖö ņøÉņ£äņäĖļć©Ļ┤ĆĻ│╝ ņ¦æĒĢ®Ļ┤ĆņŚÉ ņ£äņ╣śĒĢ£ AVP ņłśņÜ®ņ▓┤(vasopressin type 2 receptor, V2R)ņŚÉ Ļ▓░ĒĢ®ĒĢśņŚ¼ ņäĖĒżļé┤ 3ŌĆÖ-5ŌĆÖ-cyclic adenosine monophosphate (cAMP)ļź╝ ņ”ØĻ░Ćņŗ£ņ╝£ ļéŁņóģņØś ņä▒ņןņØä ņĢ╝ĻĖ░ĒĢ£ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż. ļö░ļØ╝ņä£ ļŗżļ¤ēņØś ņłśļČäņäŁņĘ©ļŖö ņŗĀņןņŚÉņä£ņØś ļ░öņåīĒöäļĀłņŗĀ ņāØņä▒ņØä ņ¢ĄņĀ£ĒĢśļŖö ĒÜ©Ļ│╝ņÖĆ ĒĢ©Ļ╗ś ļéŁņóģņØś ņä▒ņןņØä ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ņ¦Ć ņĢŖņØäĻ╣ī ĒĢśļŖö ņØ┤ļĪĀņāü ņČöņĖĪņØ┤ ņ׳ņ¢┤ņÖöļŗż. ļ░öņåīĒöäļĀłņŗĀĻ│╝ Ļ┤ĆļĀ©ĒĢ£ ņ×¼ļ»Ėņ׳ļŖö ļÅÖļ¼╝ ņŗżĒŚśņ£╝ļĪ£, ļŗżļéŁņŗĀ ļ¦łņÜ░ņŖżņØś ņłśļČäņäŁņĘ©ļź╝ ļŖśļĀĖļŹöļŗł Ēśłņżæ ļ░öņåīĒöäļĀłņŗĀ ļåŹļÅäĻ░Ć Ļ░ÉņåīļÉśļ®┤ņä£ AVP ņłśņÜ®ņ▓┤ ĻĖĖĒĢŁņĀ£ ņ╣śļŻīņÖĆ ļ╣äņŖĘĒĢśĻ▓ī ļéŁņóģ ņä▒ņןņØä ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ņŚłļŗż[18]. ĒĢ£ ņĀäļ¼ĖĻ░Ć ļ”¼ļĘ░ņŚÉņä£ļŖö ĒĢśļŻ© 2.5-4 L ņĀĢļÅä ņåīļ│ĆņØä ļ│╝ ņłś ņ׳Ļ▓ī ņłśļČäņØä ņäŁņĘ©ĒĢśņŚ¼ ņåīļ│Ć ņśżņŖżļ¬░ļåŹļÅäļź╝ 250 mOsm/kg ņØ┤ĒĢśļĪ£ Ļ░Éņåīņŗ£Ēéżļ®┤, ļ░öņåīĒöäļĀłņŗĀ ļČäļ╣äļź╝ ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņ¢ĄņĀ£ĒĢ┤ņä£ ļéŁņóģ ņä▒ņןņØä ļŖ”ņČ£ ņłś ņ׳ļŗżĻ│Ā ĻČīĻ│ĀĒĢśņśĆļŗż[19].
ĻĘĖļ¤¼ļéś ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ļŗżļ¤ēņØś ņłśļČäņäŁņĘ©Ļ░Ć ņŗżņĀ£ļĪ£ ņØ┤ļĪ£ņÜ┤ ņ¦ĆņŚÉ ļīĆĒĢ┤ņä£ļŖö ĒÖĢņŗżĒĢ£ ĻĘ╝Ļ▒░ļŖö ņŚåļŗż. ļŗżļéŁņŗĀ ĒÖśņ×É 18ļ¬ģĻ│╝ 16ļ¬ģņØä Ļ░üĻ░ü ļŗżļ¤ēņäŁņĘ©ĻĄ░(high water intake group, ĒĢśļŻ© 50 mL/kg ņØ┤ņāü ļśÉļŖö 2.5-3.0 L ņäŁņĘ©)Ļ│╝ ņ×Éņ£ĀņäŁņĘ©ĻĄ░(free water intake group)ņ£╝ļĪ£ ļ¼┤ņ×æņ£ä ļ░░ņĀĢĒĢśĻ│Ā 1ļģäĻ░ä ņČöņĀü Ļ┤Ćņ░░ĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ņĄ£ĻĘ╝ ļ░£Ēæ£ļÉśņŚłļŗż. ļæÉ ĻĄ░ Ļ░äņØś ņŚ░ĻĄ¼ ņĀä TKV ļ░Å ņŗĀņןĻĖ░ļŖźņØĆ ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć ņŚåņŚłņ£╝ļéś ņåīļ│Ć ļé┤ ņåīļööņøĆ ļ░░ņČ£ļ¤ēņØĆ ļŗżļ¤ēņäŁņĘ©ĻĄ░ņØ┤ ņ£ĀņØśĒĢśĻ▓ī ļ¦ÄņĢśļŗż. ņŚ░ĻĄ¼ Ļ▓░Ļ│╝, ļŗżļ¤ēņäŁņĘ©ĻĄ░ņŚÉņä£ ņ×Éņ£ĀņäŁņĘ©ĻĄ░ņŚÉ ļ╣äĒĢ┤ Ēśłņżæ AVPļź╝ ļīĆļ│ĆĒĢśļŖö copeptin ņłśņ╣śĻ░Ć ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīļÉśņ¢┤ ņ׳ņŚłļŗż. ĻĘĖļ¤¼ļéś ņØ╝ņ░© ĒÅēĻ░Ć ņ¦ĆĒæ£ņśĆļŹś ņĮ®Ēīź ļČĆĒö╝ņÖĆ ņØ┤ņ░© ĒÅēĻ░Ć ņ¦ĆĒæ£ņśĆļŹś ņŗĀņן ĻĖ░ļŖźņØś ļ│ĆĒÖöņŚÉ ņ׳ņ¢┤ņä£ ļæÉ ĻĄ░ ņé¼ņØ┤ņŚÉ ņ£ĀņØśĒĢ£ ņ░©ņØ┤ļŖö ņŚåņŚłļŗż. ĒĢ£ĒÄĖ, ļŗżļ¤ēņäŁņĘ©ĻĄ░ņŚÉņä£ ņØ┤ņĀäņØś ņĮ®Ēīź ļČĆĒö╝ ņ”ØĻ░ĆņÖĆ glomerular filtration rate (GFR) Ļ░Éņåī ņåŹļÅäņŚÉ ļ╣äĒĢ┤ņä£ ņŚ░ĻĄ¼ ĻĖ░Ļ░äļÅÖņĢł ņ£ĀņØśĒĢśĻ▓ī ņŗ¼ĒĢ┤ņ¦ĆļŖö Ļ▓░Ļ│╝Ļ░Ć ļéśĒāĆļé¼ļŗż[20]. ļŗżļ¤ēņäŁņĘ©ĻĄ░ņŚÉņä£ ņÜö ņåīļööņøĆ ļ░░ņČ£ļ¤ēņØ┤ ļ¦ÄņĢśĻ│Ā, ļŗżļ¤ēņØś ņłśļČäņäŁņĘ©Ļ░Ć ļ░öņåīĒöäļĀłņŗĀņŚÉ ņØśĒĢ£ ļ│┤ņāü ņ×æņÜ®ņØä ļ░®ĒĢ┤ĒĢ┤ņä£ ņ┤łĻĖ░ņŚÉ GFR Ļ░ÉņåīĒÅŁņØ┤ ņ╗ĖņØä Ļ░ĆļŖźņä▒ņØ┤ ņ׳ņ£╝ļéś[21], ņĀüņØĆ ņłśņØś ļīĆņāüņ×ÉņŚÉņä£ 1ļģäņØś ņČöņĀü Ļ┤Ćņ░░ļ¦īņ£╝ļĪ£ļŖö Ļ▓░ļĪĀņØä ņ¦ōĻĖ░ ņ¢┤ļĀĄļŗż. ļśÉ ļŗżļźĖ ņŚ░ĻĄ¼ņŚÉņä£ ļ│┤ļ®┤, 1ņŻ╝ņØ╝ Ļ░ä ĒĢśļŻ© 3 LņØś ņłśļČäņØä ņäŁņĘ©Ē¢łņØä ļĢī ņÜö ņśżņŖżļ¬░ļåŹļÅäļź╝ ĒÅēĻĘĀ 270 mOsm/kgĻ╣īņ¦Ć ļé«ņČ£ ņłś ņ׳ņŚłņ£╝ļéś, Ēśłņżæ ļ░öņåīĒöäļĀłņŗĀ ĒÖ£ņä▒ļÅäļź╝ ļ░śņśüĒĢ£ļŗżĻ│Ā ņāØĻ░üĒĢśļŖö ņÜö cAMPļŖö ļČĆļČäņĀüņ£╝ļĪ£ļ¦ī ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ņŚłļŗż[22].
Ēśäņ×¼ļĪ£ņä£ ņĀüņĀłĒĢ£ ņłśļČä ņäŁņĘ©ļŖö ļŗżļéŁņŗĀņŚÉ ĒØöĒĢ£ ņÜöļĪ£ Ļ▓░ņäØĻ│╝ Ļ░ÉņŚ╝ņØä ņśłļ░®ĒĢśļŖö ļŹ░ļÅä ļÅäņøĆņØ┤ ļÉśļ»ĆļĪ£, ņĮ®Ēīź ĻĖ░ļŖźņØ┤ ņĀĢņāüņØĖ ĒÖśņ×ÉņŚÉņä£ ņĢĮļ¼╝ņØä ņé¼ņÜ®ĒĢĀ ņłś ņ׳ĻĖ░ ņĀäĻ╣īņ¦Ć ņŗ£ļÅäĒĢ┤ ļ│╝ ļ¦īĒĢ£ ļ░®ļ▓ĢņØ┤ļØ╝Ļ│Ā ņāØĻ░üļÉ£ļŗż. ļŗżļ¤ēņØś ņłśļČä ņäŁņĘ©Ļ░Ć ļŗżļéŁņŗĀņØś ņ¦łļ│æĻ▓ĮĻ│╝ņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņŚÉ ļīĆĒĢ┤ņä£ļŖö ļīĆĻĘ£ļ¬©ņØś ņ×äņāüņŗ£ĒŚśņŚÉņä£ ĻĘ£ļ¬ģļÉśņ¢┤ņĢ╝ ĒĢ£ļŗż.
ņĀüĻĘ╣ņĀüņØĖ ĒśłņĢĢņĪ░ņĀł
Ļ│ĀĒśłņĢĢņØĆ ļŗżļéŁņŗĀņŚÉņä£ Ļ░Ćņן ĒØöĒĢ£ ņŗĀņן ņ”Øņāüņ£╝ļĪ£ ņĢĮ 65-93%ņŚÉņä£ ĒĢ®ļ│æļÉśļ®░, ņŗĀļČĆņĀä ņ¦äĒ¢ē ļ░Å ņŗ¼ĒśłĻ┤ĆĻ│ä ĒĢ®ļ│æņ”ØņØś ņŻ╝ņÜö ņ£äĒŚśņØĖņ×ÉņØ┤ļŗż. ņāüļīĆņĀüņ£╝ļĪ£ ņĀŖņØĆ ļéśņØ┤ļČĆĒä░ ĒśłņĢĢņØ┤ ņāüņŖ╣ĒĢśļŖöļŹ░, ņ¦łļ│æ ņ┤łĻĖ░ ļŗ©Ļ│äļČĆĒä░ ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│äĻ░Ć ĒÖ£ņä▒ĒÖöļÉśļŖö Ļ▓āĻ│╝ ņŚ░Ļ┤ĆļÉśņ¢┤ ņ׳ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż[23]. 75ļ¬ģņØś ĒÖśņ×Éļź╝ 7ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ ĒśłņĢĢ 120/80 mmHg ņØ┤ĒĢśļź╝ ļ¬®Ēæ£ļĪ£ ņĪ░ņĀłĒĢ£ ĻĄ░ņŚÉņä£ ņóīņŗ¼ņŗż ļ╣äļīĆ Ļ░ÉņåīĒÅŁņØ┤ ņ╗Ėņ£╝ļ®░[24], modification of diet in renal disease (MDRD) ņŚ░ņןņČöņĀüņŚ░ĻĄ¼ņŚÉ ĒżĒĢ©ļÉ£ 200ļ¬ģņØś ļŗżļéŁņŗĀ ĒÖśņ×ÉĻĄ░ņŚÉņä£ ļ¬®Ēæ£ ĒśłņĢĢņØä ļé«Ļ▓ī Ē¢łļŹś Ļ▓ĮņÜ░(ĒÅēĻĘĀ ļÅÖļ¦źņĢĢ < 92 vs. < 107 mmHg) ņŗĀļČĆņĀä ņ¦äĒ¢ēņØä ļŖ”ņČöļŖö ļŹ░ ņ£ĀņØśĒĢ£ ņśüĒ¢źņØä ļ»Ėņ│żļŹś ņĀÉņØä ĻĘ╝Ļ▒░ļĪ£ ĒĢśņŚ¼[25], < 130/80 mmHgļź╝ ļ¬®Ēæ£ļĪ£ ĒĢśņŚ¼ ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│ä ņ░©ļŗ©ņĀ£(ņĢłņ¦ĆņśżĒģÉņŗĀ ņĀäĒÖśĒÜ©ņåī ņ¢ĄņĀ£ņĀ£[angiotensin converting enzyme inhibitors, ACEi] ļśÉļŖö ņĢłņ¦ĆņśżĒģÉņŗĀ ņłśņÜ®ņ▓┤ ĻĖĖĒĢŁņĀ£ [angiotensin receptor blockers, ARB])ļź╝ ņÜ░ņäĀ ņé¼ņÜ®ĒĢśĻ│Ā ņ׳ļŗż. ĻĘĖļ¤¼ļéś ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś ĒśłņĢĢņĪ░ņĀłņŚÉ ņ׳ņ¢┤ ņĀüņĀłĒĢ£ ļ¬®Ēæ£ ĒśłņĢĢņØ┤ ņ¢╝ļ¦łņØĖņ¦Ć, ņ¢┤ļ¢ż Ļ│äņŚ┤ņØś ĒĢŁĻ│ĀĒśłņĢĢņĀ£ļź╝ ņé¼ņÜ®ĒĢśļŖö Ļ▓āņØ┤ ņ£Āļ”¼ĒĢ£ņ¦ĆņŚÉ ļīĆĒĢ┤ņä£ļŖö ĻĘ╝Ļ▒░Ļ░Ć ļČĆņĪ▒ĒĢ£ ņāüĒā£ņśĆļŗż.
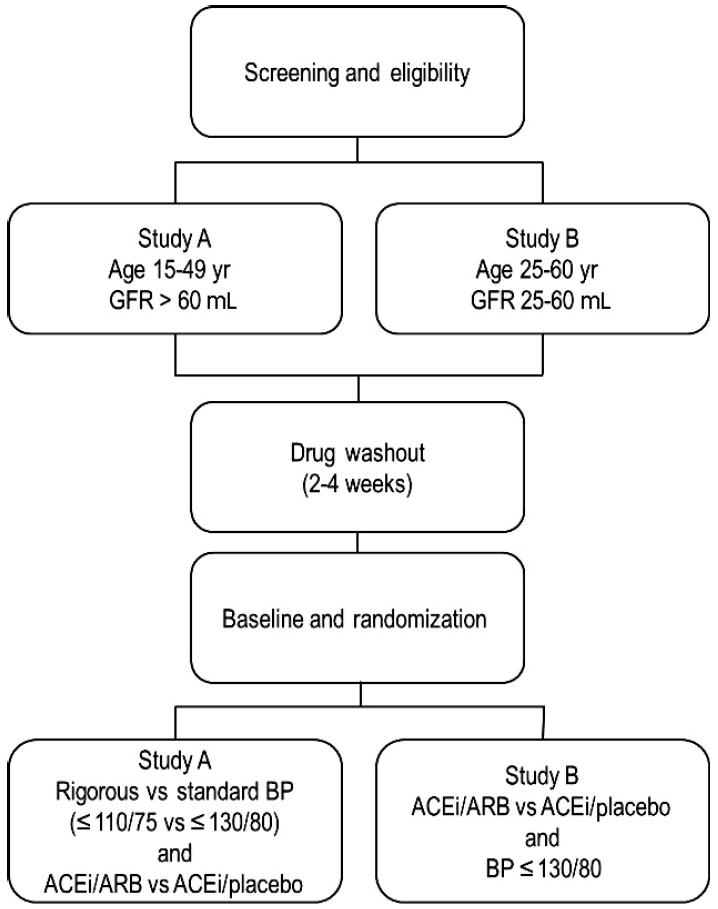
ņØ┤ņŚÉ ļīĆĒĢ£ ĒĢ┤ļŗĄņØä ņ¢╗Ļ│Āņ×É ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ņŗ£Ē¢ēĒĢ£ ļīĆĻĘ£ļ¬© ņ×äņāüņŗ£ĒŚś Ļ▓░Ļ│╝Ļ░Ć ņĄ£ĻĘ╝ņŚÉ ļ░£Ēæ£ļÉśņŚłļŗż. HALT progression of polycystic kidney disease (HALT PKD; NCT 00283686 & NCT 01885559) ņŚ░ĻĄ¼ļŖö Ļ│ĀĒśłņĢĢņØ┤ ņ׳ļŖö ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ 2006ļģä ļ»ĖĻĄŁ ĻĄŁļ”Į ļ│┤Ļ▒┤ņøÉņØś ĒøäņøÉņ£╝ļĪ£ ņŗ£ņ×æļÉśņ¢┤ 5ļģäĻ░ä ņ¦äĒ¢ēļÉśņŚłļŗż[26-28]. ņŚ░ĻĄ¼ ļīĆņāüņ×ÉņØś ņ┤łĻĖ░ ņŗĀĻĖ░ļŖźņŚÉ ļö░ļØ╝ņä£ Study AņÖĆ BļĪ£ ļéśļłäņ¢┤ ņ¦äĒ¢ēĒĢśņśĆĻ│Ā(Fig. 1), Study AņŚÉņä£ļŖö ļŗżņŗ£ ļæÉ Ļ░Ćņ¦Ć ļ¬®Ēæ£ ĒśłņĢĢ(110/75 vs 130/80 mmHg)Ļ│╝ ACEi/ARB ļ│æņÜ® ļīĆ ACEi ļŗ©ļÅģņØś ļ╣äĻĄÉļź╝ ĒĢśļŖö ņ┤Ø 4Ļ░Ćņ¦Ć ĻĄ░ņ£╝ļĪ£ ļ¼┤ņ×æņ£ä ļ░░ņĀĢĒ¢łĻ│Ā, Study BņŚÉņä£ļŖö 130/80 mmHg ņØ┤ĒĢśļĪ£ ņĪ░ņĀłĒĢśļ®┤ņä£ ACEi/ARB ļ│æņÜ® ļīĆ ACEi ļŗ©ļÅģ ņÜöļ▓ĢņØä ļ╣äĻĄÉĒ¢łļŗż. Study AļŖö ņØ╝ņ░© ĒÅēĻ░Ćņ¦ĆĒæ£ļĪ£ TKVņØś ļ│ĆĒÖöņ£©ņØä ņØ╝ņ░©ĒÅēĻ░Ćņ¦ĆĒæ£ļĪ£ ņäżņĀĢĒĢśņśĆĻ│Ā, Study BļŖö GFRņØś 50% Ļ░Éņåī, ļ¦ÉĻĖ░ņŗĀļČĆņĀä ļśÉļŖö ņé¼ļ¦ØņØś ļ│ĄĒĢ®ņØä ņØ╝ņ░© ĒÅēĻ░Ćņ¦ĆĒæ£ļĪ£ ņĢĮ 5ļģäņØä ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆļŗż.
ņÜ░ņäĀ, ņ┤łĻĖ░ ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ Ē¢łļŹś HALT AņŚ░ĻĄ¼ļŖö ņ┤Ø 558ļ¬ģņØ┤ ņ░ĖņŚ¼Ē¢łņ£╝ļ®░ Ēæ£ņżĆļ¬®Ēæ£ĒśłņĢĢĻĄ░(120/70-130/80 mmHg)Ļ│╝ ļé«ņØĆļ¬®Ēæ£ĒśłņĢĢĻĄ░(95/60-110/75 mmHg) ļ¬©ļæÉ ĒśłņĢĢņØĆ ļ¬®Ēæ£ņŚÉ ļ¦×Ļ▓ī ņ£Āņ¦ĆļÉśņŚłļŗż. Ļ▓░Ļ│╝ ņ¦ĆĒæ£ņØĖ TKV ņ”ØĻ░Ćņ£©ņØĆ Ēæ£ņżĆļ¬®Ēæ£ĒśłņĢĢĻĄ░ņŚÉ ļ╣äĒĢ┤ņä£ ļé«ņØĆļ¬®Ēæ£ĒśłņĢĢĻĄ░ņŚÉņä£ ņŚ░Ļ░ä 1% ļé«ņĢśļŗż(ņŚ░Ļ░ä 5.6 vs. 6.6%, p = 0.006). ļśÉĒĢ£, ņŗ¼ĒśłĻ┤ĆĻ│ä ĒĢ®ļ│æņ”ØņØä ļ░śņśüĒĢśļŖö ņóīņŗ¼ņŗż ņ¦łļ¤ēņ¦Ćņłś(left ventricular mass index)ļÅä ļé«ņØĆļ¬®Ēæ£ĒśłņĢĢĻĄ░ņŚÉņä£ ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīĒĢśņśĆņ£╝ļ®░(ņŚ░Ļ░ä -1.17 vs -0.57 g/m2, p < 0.001), ļŗ©ļ░▒ļć©ļÅä ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīĒĢśļŖö Ļ▓░Ļ│╝ļź╝ ļ│┤ņśĆļŗż(ņŚ░Ļ░ä -3.77 vs +2.43%, p < 0.001). ĻĘĖļ¤¼ļéś GFRņØś ļ│ĆĒÖöļŖö ļæÉ ĻĄ░ ļ¬©ļæÉ ņŚ░Ļ░ä ņĢĮ 3 mL/min/1.73 m2Ļ░ÉņåīĒĢśņŚ¼ ņ░©ņØ┤Ļ░Ć ņŚåņŚłļŗż. ACEi/ARBĻĄ░Ļ│╝ ACEi ļŗ©ļÅģĻĄ░ņØś ļ╣äĻĄÉ Ļ▓░Ļ│╝ ļæÉ Ļ░Ćņ¦Ć Ļ▓░Ļ│╝ ņ¦ĆĒæ£ ļ¬©ļæÉ ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć ņŚåņŚłļŗż. ĒĢ£ĒÄĖ, ņ¦äĒ¢ēļÉ£ ļŗ©Ļ│äņØś ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ HALT B ņŚ░ĻĄ¼ņŚÉņä£ļŖö ACEi/ARBĻĄ░Ļ│╝ ACEi ļŗ©ļÅģĻĄ░ņŚÉņä£ ĒśłņĢĢņØś ņĀĢļÅäļéś ļ│ĄĒĢ® ņ¦ĆĒæ£ņŚÉ ļīĆĒĢ£ ņāØņĪ┤ļČäņäØ Ļ▓░Ļ│╝ņŚÉņä£ ļæÉ ĻĄ░ Ļ░äņŚÉ ņ░©ņØ┤Ļ░Ć ņŚåĻ│Ā, GFR Ļ░ÉņåīņåŹļÅä ņŚŁņŗ£ ļŗżļź┤ņ¦Ć ņĢŖņĢśļŗż.
ņØ┤ņāüņØś Ļ▓░Ļ│╝ļź╝ ņóģĒĢ®ĒĢśņ×Éļ®┤, ņ┤łĻĖ░ ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ĒśłņĢĢņØś ņĀüĻĘ╣ņĀüņØĖ ņĪ░ņĀłņØĆ 1) ņŗ¼ĒśłĻ┤ĆĻ│ä ĒĢ®ļ│æņ”Ø ļ░Å ļŗ©ļ░▒ļć© Ļ░ÉņåīņØś ĒÜ©Ļ│╝Ļ░Ć ņ׳ņŚłĻ│Ā, 2) ņ┤ØņŗĀņןļČĆĒö╝ņØś ņ”ØĻ░ĆņåŹļÅäļź╝ Ļ░Éņåīņŗ£ņ╝░ņ£╝ļéś, 3) ACEi/ARB ļ│æņÜ®ņÜöļ▓ĢņØĆ ņČöĻ░ĆņĀüņØĖ ņØ┤ļōØņØĆ ņŚåņŚłļŗż. ĻĘĖļ¤¼ļéś ņĀüĻĘ╣ņĀüņØĖ ĒśłņĢĢņĪ░ņĀł ļ░Å ĒśłņĢĢņĢĮ ņĀ£ņ×¼ņØś ņóģļźśņŚÉ ļö░ļźĖ ņןĻĖ░ņĀüņØĖ ņŗĀĻĖ░ļŖź ļ│┤ņĪ┤ĒÜ©Ļ│╝ņŚÉ ļīĆĒĢ┤ņä£ļŖö ņČöĻ░ĆņĀüņØĖ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢ£ ļČĆļČäņØ┤ļŗż.
ļö░ļØ╝ņä£ ĒśłņĢĢņØĆ Ēśäņ×¼ ņČöņ▓£ļÉśļŖö 130/80 mmHg ņØ┤ĒĢśļĪ£ ņĪ░ņĀłĒĢśļÅäļĪØ ĻČīĻ│ĀĒĢśļÉś ĒÖśņ×ÉņŚÉ ļö░ļØ╝ņä£ ļ¬®Ēæ£ļź╝ Ļ░£ļ│äĒÖöĒĢ┤ņĢ╝Ļ▓Āļŗż. ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│ä ņ░©ļŗ©ņĀ£ļĪ£ ņĪ░ņĀłņØ┤ ņĢł ļÉĀ Ļ▓ĮņÜ░ ļŗżļźĖ ņóģļźśņØś ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│ä ņ░©ļŗ©ņĀ£ļź╝ ņČöĻ░ĆĒĢśĻĖ░ļ│┤ļŗżļŖö ļŗżļźĖ Ļ│äņŚ┤ņØś ĒĢŁĻ│ĀĒśłņĢĢņĀ£ļź╝ ņé¼ņÜ®ĒĢśļŖö Ļ▓ī ļ░öļ×īņ¦üĒĢśļŗżĻ│Ā ņāØĻ░üļÉśļ®░, ļÅÖļ░śņ¦łĒÖśņØ┤ļéś ņ¦łļ│æņØś ņ¦äĒ¢ē ņĀĢļÅäņŚÉ ļö░ļØ╝ ņĢĮņĀ£ļź╝ ņäĀĒāØĒĢśļŖö Ļ▓āņØ┤ ņóŗĻ▓Āļŗż. ņĀĆņ×ÉņØś Ļ▓ĮņÜ░, ņØ┤ņ░© ņĢĮņĀ£ļĪ£ ņÜ░ņäĀ ļ▓ĀĒāĆ ņ░©ļŗ©ņĀ£ ļśÉļŖö ņĢīĒīī-/ļ▓ĀĒāĆ-ņ░©ļŗ©ņĀ£(ņśł, ņ╣┤ļ▓ĀļööļĪż[carvedilol])ļź╝ ņČöĻ░ĆĒĢśĻ│Ā, ļööĒĢśņØ┤ļō£ļĪ£Ēö╝ ļ”¼ļöśĻ│ä ņ╣╝ņŖśĒåĄļĪ£ņ░©ļŗ©ņĀ£ļź╝ ļŗżņØīņ£╝ļĪ£ ņé¼ņÜ®ĒĢ£ļŗż. ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś ļīĆļČĆļČäņØĆ ņÜöļ╣äņżæņØ┤ ļé«Ļ│Ā, ļŗżļć©(polyuria)Ļ░Ć ĒØöĒĢ£ ĒŖ╣ņä▒ņØ┤ ņ׳ņ¢┤ņä£ ņØ┤ļć©ņĀ£Ļ░Ć ĒĢäņÜöĒĢ£ Ļ▓ĮņÜ░Ļ░Ć ļ¦Äņ¦Ć ņĢŖĻ│Ā, ņØ┤ļć©ņĀ£ļŖö ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│äļź╝ ĒÖ£ņä▒ĒÖöĒĢśĻ▒░ļéś, AVP ņ”ØĻ░Ć ļ░Å cAMPļź╝ ĒÖ£ņä▒ĒÖöĒĢĀ ņłś ņ׳ļŗżļŖö ņĀÉņŚÉ ņ£ĀņØśĒĢ┤ņĢ╝ ĒĢ£ļŗż. ļśÉĒĢ£, ļ▓ĀļØ╝Ēīīļ░Ć(verapamil) Ļ░ÖņØĆ ļ╣ä-ļööĒĢśņØ┤ļō£ļĪ£Ēö╝ļ”¼ļöśĻ│ä ņ╣╝ņŖśĒåĄļĪ£ņ░©ļŗ©ņĀ£ļŖö ļéŁņóģņä▒ņןņØä ņĢģĒÖöņŗ£Ēé©ļŗżļŖö ļÅÖļ¼╝ņŗżĒŚś ļ│┤Ļ│ĀĻ░Ć ņ׳ņ£╝ļ»ĆļĪ£ Ēö╝ĒĢśļŖö Ļ▓āņØ┤ ņóŗĻ▓Āļŗż[29].
ĒĢ®ļ│æņ”Ø Ļ┤Ćļ”¼
Ēśłļć© ļ░Å ļéŁņóģņČ£Ēśł
ļéŁņóģ ņČ£Ēśł ļ░Å ņ£ĪņĢłņĀü Ēśłļć©ļŖö ļŗżļéŁņŗĀņŚÉņä£ ĒØöĒ׳ ļÅÖļ░śļÉśļŖö ĒĢ®ļ│æņ”Øņ£╝ļĪ£, ņĢĮ 30-50%ņŚÉņä£ ļÅÖļ░śļÉśļ®░, ļīĆĻ░£ ĒĢ£ ĒÖśņ×ÉņŚÉņä£ļÅä ļ░śļ│ĄņĀüņ£╝ļĪ£ ļ░£ņāØĒĢ£ļŗż[30]. Ēśłļć©ņØś ĒØöĒĢ£ ņøÉņØĖņ£╝ļĪ£ļŖö ļéŁņóģ ņČ£Ēśł, ņÜöļĪ£ Ļ▓░ņäØ, ļéŁņóģ Ļ░ÉņŚ╝ ļō▒ņØ┤ ņ׳ņ£╝ļ®░, ĒØöĒĢśņ¦Ć ņĢŖĻ▓īļŖö ņóģņ¢æņØ┤ ņ׳ņØä ņłś ņ׳ļŗż. ļéŁņóģ ņČ£Ēśłļ¦īņ£╝ļĪ£ļÅä ļ░£ņŚ┤ņØ┤ ļÉĀ ņłś ņ׳ņ£╝ļ®░, ņØ┤ļĢīņŚÉļŖö ļéŁņóģ Ļ░ÉņŚ╝Ļ│╝ņØś ĻĄ¼ļČäņØ┤ ņ¢┤ļĀżņÜĖ ņłś ņ׳ļŗż. ņ£ĪņĢłņĀü Ēśłļć©Ļ░Ć ļ░£ņāØĒĢ£ Ļ▓ĮņÜ░ņŚÉļŖö ņČ®ļČäĒĢ£ ņłśļČä ņäŁņĘ©, ņ╣©ņāü ņĢłņĀĢ, ĒåĄņ”Ø ņĪ░ņĀłņØä ĒĢśļ®┤ ļīĆĻ░£ 2-7ņØ╝ ļé┤ ņ”ØņāüņØ┤ ņÖäĒÖöļÉ£ļŗż. ĻĘĖļ¤¼ļéś ļ│┤ņĪ┤ņĀü ņÜöļ▓ĢņŚÉļÅä 7ņØ╝ ņØ┤ņāü ņ¦ĆņåŹļÉśļŖö ņ£ĪņĢłņĀü Ēśłļć©Ļ░Ć ņ׳Ļ▒░ļéś, Ļ│ĀņŚ┤ ļō▒ ļéŁņóģ Ļ░ÉņŚ╝ņØś ĒĢ®ļ│æņØ┤ ņØśņŗ¼ļÉĀ ļĢī, ĒśłņĢĢņØ┤ Ļ░ÉņåīļÉĀ ņĀĢļÅäļĪ£ ņŗ¼ĒĢ£ ņČ£ĒśłņØ┤ ņØśņŗ¼ļÉśļŖö Ļ▓ĮņÜ░ņŚÉļŖö ņØśļŻīņ¦äņŚÉĻ▓ī ļ¼ĖņØśĒĢśņŚ¼ ņ×ģņøÉņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢĀ ņłś ņ׳ļŗż. ļéŁņóģ ņČ£ĒśłņØś ņ┤łĻĖ░ņŚÉļŖö ĻĖēņä▒ ņŗĀļČĆņĀäņØś ņ£äĒŚśņä▒ ļĢīļ¼ĖņŚÉ ļĀłļŗī-ņĢłņ¦ĆņśżĒģÉņŗĀĻ│ä ņ░©ļŗ©ņĀ£ļéś ņØ┤ļć©ņĀ£, ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£ņØś ņé¼ņÜ® ņżæļŗ©ņØ┤ ĻČīĻ│ĀļÉ£ļŗż[31]. ņ”ØļĪĆ ļ│┤Ļ│Ā ņłśņżĆņØ┤ĻĖ┤ ĒĢśņ¦Ćļ¦ī ļ│┤ņĪ┤ņĀü ņÜöļ▓ĢņŚÉļÅä ļōŻņ¦Ć ņĢŖļŖö ļ¦īņä▒ņĀüņØĖ Ēśłļć©ļéś ņČ£ĒśłņØ┤ ņ׳ņØä Ļ▓ĮņÜ░ ĒŖĖļØ╝ļäźņé¼ļ»╝ņé░(tranexamic acid)ņØä ņé¼ņÜ®ĒĢ┤ņä£ ĒÜ©Ļ│╝ņĀüņ£╝ļĪ£ ņČ£ĒśłņØä ņ╣śļŻīĒĢ£ Ļ▓ĮņÜ░Ļ░Ć ļ│┤Ļ│ĀļÉśņŚłļŗż[32,33].
ņÜöļĪ£ Ļ▓░ņäØ
ņÜöļĪ£ Ļ▓░ņäØņØĆ ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś 20%ņŚÉņä£ Ļ┤Ćņ░░ļÉśļ®░, ņŗĀņןļé┤ ņåīļ│Ć ņĀĢņ▓┤ņÖĆ ņÜöņżæ citrateņØś Ļ░ÉņåīĻ░Ć ļ░£ļ│æņØś ņŻ╝ņÜöĻĖ░ņĀäņØ┤ļŗż. ņÜöņé░ Ļ▓░ņäØņØ┤ ņ╣╝ņŖś Ļ▓░ņäØļ│┤ļŗż ļŹö ļ╣łļ▓łĒĢ£ļŹ░, ņØ┤ļŖö ņĢöļ¬©ļŗłņĢä ļ░░ņäż ņןņĢĀņŚÉ ļö░ļźĖ ņÜö ņé░ņä▒ĒÖöņŚÉ ņØśĒĢ£ Ļ▓āņ£╝ļĪ£ ĒĢ┤ņäØļÉ£ļŗż[34]. ņÜöļĪ£Ļ▓░ņäØņØĆ ļ╣äņĪ░ņśü CTļĪ£ ņ¦äļŗ©ĒĢśļ®░, ņĄ£ĻĘ╝ ļōĆņ¢╝ņŚÉļäłņ¦Ć(dual energy) CTļĪ£ ņÜöņé░Ļ▓░ņäØņØä ņ╣╝ņŖśĻ▓░ņäØņ£╝ļĪ£ļČĆĒä░ ĻĄ¼ļČäĒĢ┤ ļé╝ ņłś ņ׳ļŗżļŖö ļ│┤Ļ│ĀĻ░Ć ļéśņśżĻ│Ā ņ׳ļŗż[35]. ņÜöņé░Ļ▓░ņäØ ļ░Å ņłśņé░ĒÖöņ╣╝ņŖś(calcium oxalate) Ļ▓░ņäØņØś Ļ▓ĮņÜ░ ĻĄ¼ņŚ░ņé░ņ╣╝ļź©(potassium citrate) ņ╣śļŻīĻ░Ć ĻČīņןļÉ£ļŗż. ņÜöņé░Ļ▓░ņäØņØ┤ ņ׳ļŖö ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ņÜöņé░ņØä ļé«ņČöļŖö ņ╣śļŻīļź╝ ĒĢśļŖö Ļ▓āņØ┤ Ļ▓░ņäØņØś ļ╣łļÅäļź╝ ņżäņØĖļŗżļŖö ļ│┤Ļ│ĀļŖö ņĢäņ¦ü ņŚåļŗż. ņ▓┤ņÖĖņČ®Ļ▓®ĒīīņćäņäØņłĀ(extracorporeal shock wave lithotripsy)ņØ┤ļéś Ļ▓ĮĒö╝ņĀü ņŗĀņĀłņäØņłĀ(percutaneous nephrostolithotomy) ļō▒ņØĆ Ļ▓ĮĒŚśņ×ÉņŚÉ ņØśĒĢ┤ ņŗ£ļÅäļÉĀ ļĢī ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ļÅä ņĢłņĀäĒĢśĻ▓ī ņŗ£ļÅäļÉĀ ņłś ņ׳ļŗż[36].
ļéŁņóģ Ļ░ÉņŚ╝
ļéŁņóģ Ļ░ÉņŚ╝ņØä ĒżĒĢ©ĒĢ£ ņÜöļĪ£Ļ░ÉņŚ╝ņØĆ ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś 20-25%ņŚÉņä£ ĒÅēņāØ 1ļ▓ł ņØ┤ņāü Ļ▓ĮĒŚśĒĢ£ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░, ļéŁņóģ Ļ░ÉņŚ╝(ņŗĀņן ļéŁņóģ ļ░Å Ļ░äļéŁņóģ Ļ░ÉņŚ╝)ņØä Ļ░Éļ│äņ¦äļŗ©ĒĢśļŖö Ļ▓āņØĆ ļ¦żņÜ░ ņ¢┤ļĀĄļŗż. ĒÖĢņ¦äņØĆ ņØśņŗ¼ļÉśļŖö ļéŁņóģņŚÉņä£ ļéŁņóģņĢĪņØä ņČöņČ£ĒĢśņŚ¼ ĻĘĀņØä ļÅÖņĀĢĒĢśĻ▒░ļéś ņŚ╝ņ”ØņåīĻ▓¼ņØä Ļ┤Ćņ░░ĒĢśļŖö Ļ▓āņØ┤ņ¦Ćļ¦ī, ņŗżņĀ£ļĪ£ ļŗżņłśņØś ļéŁņóģņØ┤ ņ׳ļŖö ņŗĀņןņŚÉņä£ Ļ░ÉņŚ╝ļÉ£ ļéŁņóģņØä ņ░ŠĻĖ░ļÅä ņ¢┤ļĀĄĻ│Ā ļéŁņóģņĢĪņØä ņČöņČ£ĒĢśĻĖ░ļÅä ņ¢┤ļĀżņÜ░ļ»ĆļĪ£ ļīĆĻ░£ļŖö ņ×äņāüņĀüņ£╝ļĪ£ ņ¦äļŗ©ĒĢśĻ│Ā ņ׳ļŗż. 1) ļ░£ņŚ┤ņØ┤ 38.5Ōäā ņØ┤ņāü 3ņØ╝Ļ░ä ņ¦ĆņåŹļÉśļ®┤ņä£ 2) ļ│ĄļČĆ ĒåĄņ”Ø(ļīĆĻ░£ļŖö ĒŖ╣ņĀĢļČĆņ£äļź╝ Ļ░Ćļ”¼Ēé¼ ņłś ņ׳ņØī)ņØ┤ ļÅÖļ░śļÉśĻ│Ā 3) C-reactive proteinņØ┤ 50 mg/L ņØ┤ņāü ņāüņŖ╣ļÉśņ¢┤ ņ׳ņ£╝ļ®┤ņä£, ļéŁņóģ ņČ£ĒśłņØä ļ░░ņĀ£ĒĢĀ ņłś ņ׳ņØä ļĢī ņ×äņāüņĀüņ£╝ļĪ£ ļéŁņóģĻ░ÉņŚ╝ņØä ņ¦äļŗ©ĒĢĀ ņłś ņ׳ļŗż[37]. ņĄ£ĻĘ╝ņŚÉļŖö ņ¢æņĀäņ×É ļŗ©ņĖĄņ┤¼ņśü( 18F-fluorodeoxyglucose-positron emission tomography)ņØ┤ ļéŁņóģĻ░ÉņŚ╝ņØä ņ¦äļŗ©ĒĢśļŖö ļŹ░ Ļ░Ćņן ĒÜ©Ļ│╝ņĀüņØĖ ņśüņāü Ļ▓Ćņé¼ļĪ£ ļČĆĻ░üļÉśĻ│Ā ņ׳ļŗż[38]. ņ╣śļŻīļŖö ļéŁņóģ Ēł¼Ļ│╝ļĀźņØ┤ ņóŗņØĆ fluoroquinolone Ļ│äņŚ┤ņØä ņØ╝ņ░© Ļ▓ĮĒŚśņĀü ĒĢŁņāØņĀ£ļĪ£ ņé¼ņÜ®ĒĢśļ®░, ļ░░ņ¢æ Ļ▓░Ļ│╝ņÖĆ ņ╣śļŻī ļ░śņØæņŚÉ ļö░ļØ╝ņä£ ļ│ĆĻ▓ĮĒĢ£ļŗż. ļīĆĻ░£ 4-6ņŻ╝Ļ░ä ĒĢŁņāØņĀ£ļź╝ ņ£Āņ¦ĆĒĢśļÅäļĪØ ĻČīĻ│ĀĒĢśņ¦Ćļ¦ī, ņĢäņ¦ü ņĀüņĀłĒĢ£ ĒĢŁņāØņĀ£ ņé¼ņÜ®ĻĖ░Ļ░äņØ┤ļéś ņ╣śļŻīļ░śņØæņØś ĒÅēĻ░Ćņŗ£ĻĖ░, ĻĘĖļ”¼Ļ│Ā ņłśņłĀņĀü ļ░░ņĢĪņØ┤ ĒĢäņÜöĒĢ£ ņŗ£ĻĖ░ņŚÉ ļīĆĒĢ┤ņä£ļŖö ļŹö ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśļŗż.
ļ¦īņä▒ ĒåĄņ”Ø
ļŗżļéŁņŗĀņŚÉņä£ņØś ļ¦īņä▒ ņśåĻĄ¼ļ”¼ ļśÉļŖö ĒŚłļ”¼ĒåĄņ”ØņØĆ ņŗĀņן ļśÉļŖö Ļ░äņØś ĻĖēņåŹĒĢ£ ļéŁņóģņØś ņä▒ņןĻ│╝ Ļ┤ĆļĀ©ļÉ£ ņŗĀņןņä▒ĒåĄņ”ØĻ│╝ ļéŁņóģņØś ņä▒ņןĻ│╝ļŖö ņĀäĒśĆ ņØ╝ņ╣śĒĢśņ¦Ć ņĢŖļŖö ļ¼╝ļ”¼ņĀüņØĖ ĒåĄņ”ØņØ┤ ņ׳ņØä ņłś ņ׳ļŗż(ņśł, ņÜöĒåĄ) [39]. ļ¦īņä▒ ņŗĀņןĒåĄņ”Ø(ņśåĻĄ¼ļ”¼ ĒåĄņ”Ø ļśÉļŖö ņÜöĒåĄ)ņØĆ ļŗżļéŁņŗĀņØś Ļ░Ćņן ĒØöĒĢ£ ņŗĀņן ņ”Øņāüņ£╝ļĪ£ ĻĖēņä▒ ĒåĄņ”ØņØ┤ ļ░£ņāØĒĢ£ Ēøä ņŗĀņןņØä ņ¦Ćļ░░ĒĢśļŖö Ļ░ÉĻ░üņŗĀĻ▓Į ļ░Å ņ×Éņ£©ņŗĀĻ▓ĮĻ│äņØś ņ¦ĆņåŹņĀüņØĖ ĒÖ£ņä▒ĒÖöļĪ£ ņØĖĒĢ┤ ņāØĻĖ┤ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż[40]. ļ¦īņä▒ņĀüņØĖ ĒåĄņ”ØņØĆ ĒÖśņ×ÉņØś ņéČņØś ņ¦łņØä ņĀĆĒĢśņŗ£ĒéżĻ│Ā, ņØ┤ļĪ£ ņØĖĒĢ£ ņ¦äĒåĄņåīņŚ╝ņĀ£ņØś ļ░śļ│ĄņĀüņØĖ ļ│ĄņÜ®ņØĆ ņŗĀņןĻĖ░ļŖźņØä ņĢģĒÖöņŗ£Ēé¼ ņłś ņ׳ņ£╝ļ»ĆļĪ£, ņĀüņĀłĒĢ£ ĒåĄņ”Øņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢśļŗż. ņĢĮļ¼╝ņ╣śļŻīļĪ£ļŖö ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£ņØś ņé¼ņÜ®ņØä Ēö╝ĒĢśĻ│Ā ņĢäņäĖĒŖĖņĢäļ»ĖļģĖĒÄ£ņ£╝ļĪ£ ņŗ£ņ×æĒĢśņŚ¼, ļŗ©ļÅģņÜöļ▓Ģņ£╝ļĪ£ ņĪ░ņĀłļÉśņ¦Ć ņĢŖņØä ļĢīņŚÉļŖö Ļ▓ĮĒĢ£ ļ¦łņĢĮņä▒ ņ¦äĒåĄņĀ£(ņśł, ĒŖĖļØ╝ļ¦łļÅī)ņØä ļ│æĒĢ®ĒĢśņŚ¼ ņé¼ņÜ®ĒĢ┤ ļ│╝ ņłś ņ׳ļŗż. ļīĆĻ░£ ļ¬©ļź┤ĒĢĆ Ļ│äņŚ┤ņØś Ļ░ĢĒĢ£ ļ¦łņĢĮņä▒ ņ¦äĒåĄņĀ£ļź╝ ņō░ļŖö Ļ▓ĮņÜ░ļŖö ļō£ļ¼╝ļŗż. ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£ļÅä ļŗ©ĻĖ░Ļ░ä ņĢäņäĖĒŖĖņĢäļ»ĖļģĖĒÄ£Ļ│╝ ļ│æĒĢ®ĒĢśņŚ¼ ņé¼ņÜ®ĒĢ┤ ļ│╝ ņłś ņ׳ņ£╝ļéś ņŗĀņןĻĖ░ļŖźņØ┤ ļ¢©ņ¢┤ņ¦ä ĒÖśņ×ÉņŚÉņä£ļŖö ņŻ╝ņØśļź╝ ņÜöĒĢ£ļŗż. ņĢĮļ¼╝ņ╣śļŻīļĪ£ļÅä ņĪ░ņĀłļÉśņ¦Ć ņĢŖļŖö ĒåĄņ”ØņØ┤ ņ׳ņØä ļĢīņŚÉļŖö ļéŁņóģ Ļ▓ĮĒÖöņÜöļ▓ĢņØ┤ļéś ļ│ĄĻ░ĢņŗĀĻ▓Įņ░©ļŗ©ņłĀ(celiac plexus blockade) ļō▒ņØ┤ ņé¼ņÜ®ļÉĀ ņłś ņ׳ļŗż[39].
ļćīļÅÖļ¦źĻĮłļ”¼
ļćīļÅÖļ¦źĻĮłļ”¼(intracranial aneurysm)ļŖö ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś 9-12%ņŚÉņä£ ļÅÖļ░śļÉśļ®░, ļćīļÅÖļ¦źĻĮłļ”¼ļéś ļćīņČ£ĒśłņØś Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳ļŖö ĒÖśņ×ÉņŚÉņä£ ļŹö ļ╣łļ▓łĒĢśĻ▓ī ņØ╝ņ¢┤ļé£ļŗż[41]. ļćīļÅÖļ¦źĻĮłļ”¼Ļ░Ć ņØ╝ļ░śņØĖĻĄ¼(2-3%)ļ│┤ļŗż ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ļŹö ļ╣łļ▓łĒĢśĻ▓ī ņØ╝ņ¢┤ļéśļ®░, ļśÉĒĢ£ ļŹö ņĪ░ĻĖ░(ļŗżļéŁņŗĀ vs ņØ╝ļ░śņØĖĻĄ¼ņ¦æļŗ©, 41 vs 51ņäĖ)ņŚÉ ĒīīņŚ┤ļÉśņ¢┤ ļćīņČ£ĒśłņØä ļ░£ņāØņŗ£Ēé¼ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņ×äņāüņĀüņ£╝ļĪ£ Ļ░Ćņן ņżæņÜöĒĢ£ ņŗĀņןņÖĖ ĒĢ®ļ│æņ”ØņØ┤ļŗż. ĻĘĖļ¤¼ļéś Ēü¼ĻĖ░Ļ░Ć ņ×æĻ▒░ļéś ņČ£ĒśłņØś ņ£äĒŚśņØä ņ¦Ćļŗłņ¦Ć ņĢŖņØĆ ļćīļÅÖļ¦źĻĮłļ”¼Ļ░Ć ļīĆļČĆļČäņØ┤ļ»ĆļĪ£, ļćīļÅÖļ¦źĻĮłļ”¼ļéś ļćīņČ£ĒśłņØś Ļ░ĆņĪ▒ļĀźņØä ņ¦ĆļģöĻ▒░ļéś, ļćīņČ£ĒśłņØś Ļ│╝Ļ▒░ļĀźņØä ņ¦Ćļŗī ĒÖśņ×Éļź╝ ņĀ£ņÖĖĒĢśĻ│ĀļŖö ļ¬©ļōĀ ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ņØś ļćīļÅÖļ¦źĻĮłļ”¼ņØś ņäĀļ│äĻ▓Ćņé¼ļŖö ĒĢäņÜöĒĢśņ¦Ć ņĢŖļŗż. ņäĀļ│äĻ▓Ćņé¼ļĪ£ļŖö ļćī magnetic resonance angiography (MRA)ļź╝ ņé¼ņÜ®ĒĢśļ®░, Ļ░ĆņĪ▒ļĀźņØ┤ ņ׳ļŖö ĒÖśņ×ÉņŚÉņä£ļŖö ņäĀļ│ä Ļ▓Ćņé¼ņŚÉņä£ ņ¢æņä▒ ņåīĻ▓¼ņØ┤ļ®┤ņä£ ļ¼┤ņ”ØņāüņØ┤ļ®┤ Ēü¼ĻĖ░ņŚÉ ļö░ļØ╝ 6-24Ļ░£ņøö Ļ░äĻ▓®ņ£╝ļĪ£ ļćī MRAļź╝ ņČöņĀüĻ┤Ćņ░░ĒĢ£ļŗż[42]. ņäĀļ│äĻ▓Ćņé¼ņŚÉņä£ ņØīņä▒ ņåīĻ▓¼ņØ┤ļ®┤ 5-10ļģä Ēøä ļćī MRAļź╝ ņ×¼ņŗ£Ē¢ēĒĢ┤ ļ│╝ Ļ▓āņØä ĻČīņ£ĀĒĢ£ļŗż. ļćīļÅÖļ¦ź ĻĮłļ”¼Ļ░Ć ĒīīņŚ┤ļÉśņŚłĻ▒░ļéś ņ”ØņāüņØä ļÅÖļ░śĒĢ£ ļćīļÅÖļ¦ź ĻĮłļ”¼ņØś Ļ▓ĮņÜ░ņŚÉļŖö ļćīļÅÖļ¦ź ĻĮłļ”¼ņØś Ļ▓ĮļČĆ(neck)ļź╝ ņÖĖĻ│╝ņĀüņ£╝ļĪ£ Ēü┤ļ”Į(clipping)ĒĢśĻ▒░ļéś ĒśłĻ┤Ć ļé┤ ņĮöņØ╝(endovascular coiling) ņ╣śļŻīļź╝ ĒĢ£ļŗż.
Ļ░äļéŁņóģ
Ļ░äļéŁņóģņØĆ ļŗżļéŁņŗĀ ĒÖśņ×ÉņØś Ļ░Ćņן ĒØöĒĢ£ ņŗĀņןņÖĖ ĒĢ®ļ│æņ”Øņ£╝ļĪ£ 80-90%ņŚÉņä£ ļÅÖļ░śļÉśņ¢┤ ņ׳ļŗż. ļīĆļČĆļČäņØĆ ņ”ØņāüņØä ņ┤łļלĒĢśņ¦Ć ņĢŖņ¦Ćļ¦ī ņĢĮ 20%ņŚÉņä£ļŖö Ļ░äļéŁņóģņØś ņä▒ņןņŚÉ ļö░ļźĖ ļ¼╝ļ”¼ņĀüņØĖ ņĢĢļ░ĢĻ░É, ĒåĄņ”Ø, Ēżļ¦īĻ░É ļō▒ņØ┤ ļ░£ņāØĒĢĀ ņłś ņ׳ņ£╝ļ®░, ĒŖ╣Ē׳ ņŚ¼ņä▒ņŚÉņä£ļŖö ļŗżņé░ļĀźņØ┤ ņ׳Ļ▒░ļéś ņŚ¼ņä▒ĒśĖļź┤ļ¬¼ņØä ļ│ĄņÜ®ĒĢ£ Ļ▓ĮņÜ░ ņŚ░ļĀ╣ņØ┤ ņ”ØĻ░ĆĒĢ©ņŚÉ ļö░ļØ╝ Ļ░äļéŁņóģņØś ņä▒ņןņØ┤ ļÜ£ļĀĘĒĢ┤ņ¦äļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż[43]. Ļ░äļéŁņóģņØś ņ╣śļŻīļŖö ņĀłļīĆņĀüņ£╝ļĪ£ ĒÖśņ×ÉņØś ņ”ØņāüņØä ņżäņØ┤ļŖö ļŹ░ ņ׳ļŗż. ņśłļ░®ņĀüņ£╝ļĪ£ļŖö Ļ▓ĮĻĄ¼ Ēö╝ņ×äņĢĮņØä ņżæļŗ©ĒĢśļÅäļĪØ ĻČīņ£ĀĒĢśĻ│Ā, ĻĖēļ¦īņä▒ ĒåĄņ”ØņŚÉ ļīĆĒĢ┤ņä£ļŖö ņĀüņĀłĒĢ£ ņ¦äĒåĄņĀ£ļź╝ ņé¼ņÜ®ĒĢ£ļŗż. ņĢĮļ¼╝ņ╣śļŻī(ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņ£Āņé¼ņ▓┤) ļśÉļŖö ņłśņłĀņĀü ņ╣śļŻī(ļéŁņóģ ļ░░ņĢĪņłĀ, Ļ▓ĮĒö╝ņĀü Ļ░äļÅÖļ¦ź ņāēņĀäņłĀ, Ļ░äņØ┤ņŗØ)ļŖö ņ”ØņāüņØ┤ ņŗ¼ĒĢ£ ņØ╝ļČĆ ĒÖśņ×ÉņŚÉņä£ļ¦ī ņŗ£ļÅäļÉśĻ│Ā ņ׳ļŗż.
Ļ│Āņ£äĒŚśĻĄ░ņŚÉņä£ ņŗ£ļÅäļÉśļŖö ņāłļĪ£ņÜ┤ ņĢĮļ¼╝ ņ╣śļŻīņĀ£
ļéŁņóģ ņäĖĒżņØś ņŗĀĒśĖņĀäļŗ¼Ļ│ä ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ ļéŁņóģ ņäĖĒżļé┤ ņ╣╝ņŖś ĒĢŁņāüņä▒ņØś ļ│ĆĒÖöņÖĆ cAMP ņ”ØĻ░ĆĻ░Ć ĒĢĄņŗ¼ņĀüņØĖ ņØ┤ņāüņ£╝ļĪ£ ņĀ£ņĢłļÉśņŚłļŗż[44]. ņØ┤ļ¤¼ĒĢ£ ļ│ĆĒÖöņØś ņØ┤ņ░© Ļ▓░Ļ│╝ļĪ£ ĒĢśļČĆņØś ņŚ¼ļ¤¼ ņŗĀĒśĖņĀäļŗ¼Ļ│äņŚÉ ļ│ĆĒÖöĻ░Ć ļéśĒāĆļéśļŖöļŹ░ ņØ┤ļź╝ ņ░©ļŗ©ĒĢśĻ▒░ļéś ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ļŖö ņĢĮļ¼╝ļōżņØ┤ ņŗ£ļÅäļÉśĻ│Ā ņ׳ņ£╝ļ®░, ņĀäņ×äņāüņŗ£ĒŚśĻ│╝ ņ×äņāüņŗ£ĒŚśņØ┤ ņ¦äĒ¢ē ņżæņØ┤ļŗż. ņØ┤ļōż ņżæ ņĄ£ĻĘ╝ ņŻ╝ņÜö Ļ▓░Ļ│╝Ļ░Ć ļ░£Ēæ£ļÉ£ ļ░öņåīĒöäļĀłņŗĀ ņłśņÜ®ņ▓┤ ĻĖĖĒĢŁņĀ£(vasopressin receptor 2 antagonist, V2RA), ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņ£Āņé¼ņ▓┤(somatostatin analogue, SA)ļź╝ ņżæņŗ¼ņ£╝ļĪ£ ņåīĻ░£ĒĢśĻ▓Āļŗż.
cAMP ņ¢ĄņĀ£
ļ░öņåīĒöäļĀłņŗĀ ņłśņÜ®ņ▓┤ ĻĖĖĒĢŁņĀ£(V2RA)
ņĮ®Ēīź ņ¦æĒĢ®Ļ┤Ć ņŻ╝ņäĖĒż(principal cell)ņØś basolateral membraneņŚÉ ņ£äņ╣śĒĢ£ V2RļŖö ļ░öņåīĒöäļĀłņŗĀĻ│╝ Ļ▓░ĒĢ®ĒĢśļ®┤ adenylyl cyclaseļź╝ ĒÖ£ņä▒ĒÖöĒĢ┤ņä£ cAMPļź╝ ņ”ØĻ░Ćņŗ£Ēé©ļŗż. ņĀäņ×äņāü ņŚ░ĻĄ¼ņŚÉņä£ cAMPļŖö ļéŁņóģņĢĪ ļČäļ╣äņÖĆ ņäĖĒż ņ”ØņŗØņØä ņ£ĀļÅäĒĢśņŚ¼ ļéŁņóģ ņä▒ņןņŚÉ Ļ┤ĆņŚ¼ĒĢ£ļŗżĻ│Ā ļ░ØĒśĆņĪīĻ│Ā, ļŗżļéŁņŗĀ ļÅÖļ¼╝ļ¬©ļŹĖĻ│╝ AVP knockout ļ¦łņÜ░ņŖżļź╝ ĻĄÉļ░░ĒĢ£ Ļ▓ĮņÜ░ ļéŁņóģ ņä▒ņןņØ┤ ĒśäņĀĆĒ׳ Ļ░ÉņåīļÉśļŖö Ļ▓░Ļ│╝ļź╝ ĒÖĢņØĖĒĢĀ ņłś ņ׳ļŗż[45]. ņØ┤ļ¤░ ļ░öņåīĒöäļĀłņŗĀņØś ņ×æņÜ®ņØä ņ░©ļŗ©ĒĢĀ ņłś ņ׳ļŖö ņĢĮļ¼╝ņØ┤ ļ▒üĒāä(vaptans)ļōżļĪ£, Ēå©ļ▒üĒāä(tolvaptan)ņØ┤ Ēśäņ×¼ ņĀĆļéśĒŖĖļź©Ēśłņ”ØņØś ņĪ░ņĀłņŚÉ ļīĆĒĢ┤ņä£ ĒŚłĻ░ĆļÉśņ¢┤ ņŗ£ĒīÉ ņżæņØ┤ļŗż. ņŚ¼ļ¤¼ ļÅÖļ¼╝ņŗżĒŚś Ļ▓░Ļ│╝ļź╝ ļ░öĒāĢņ£╝ļĪ£ 2007ļģäļČĆĒä░ Ēå©ļ▒üĒāäņØś ĒÜ©ļŖźĻ│╝ ņĢłņĀĢņä▒ņØä ĒÅēĻ░ĆĒĢśļŖö tolvaptan efficacy and safety in management of autosomal dominant polycystic kidney disease and its outcomes (TEMPO) 3/4 ņŚ░ĻĄ¼Ļ░Ć ņŗ£ņ×æļÉśņ¢┤ 2013ļģä ļ¦É ņŚ░ĻĄ¼Ļ▓░Ļ│╝Ļ░Ć ļ░£Ēæ£ļÉśņŚłļŗż[46,47]. 50ņäĖ ņØ┤ĒĢśņØś ņä▒ņØĖņ£╝ļĪ£ TKV > 750 mLņØ┤Ļ│Ā GFR > 60 mL/min (Cockcroft-Gault ņČöņĀĢņŗØ ņØ┤ņÜ®)ņŚÉ ĒĢ┤ļŗ╣ĒĢśļŖö 1,445ļ¬ģņØś ļŗżļéŁņŗĀ ĒÖśņ×ÉļōżņØ┤ ņ░ĖņŚ¼ļīĆņāüņØ┤ņŚłļŗż. Ēå©ļ▒üĒāäņØä 3ļģäĻ░ä Ēł¼ņŚ¼ĒĢśņśĆņØä ļĢī ņĮ®Ēīź ļČĆĒö╝ ņ”ØĻ░ĆĻ░Ć ņ£äņĢĮĻĄ░ņŚÉņä£ ņŚ░ 5.5%ņØĖ ļ░śļ®┤ ņ╣śļŻīĻĄ░ņŚÉņä£ 2.8%ļĪ£ ļéśĒāĆļéś ņĢĮ 50%ņØś ņ¢ĄņĀ£ ĒÜ©Ļ│╝ļź╝ ļéśĒāĆļāłļŗż. ļśÉĒĢ£ ĒåĄņ”ØĻ│╝ ņĮ®ĒīźĻĖ░ļŖź Ļ░Éņåī ņåŹļÅäļź╝ ņÖäĒÖöĒĢśņśĆļŗż. ņØ┤ļ¤░ Ļ▓░Ļ│╝ļŖö ņØ╝ļ│ĖņØĖņØä ļīĆņāüņ£╝ļĪ£ ĒĢ£ ĒĢśņ£äĻĄ░ ļČäņäØ(n = 177)ņŚÉņä£ļÅä ļ¦łņ░¼Ļ░Ćņ¦ĆņśĆļŗż[48]. ĻĘĖļ¤¼ļéś Ēå©ļ▒üĒāä Ēł¼ņŚ¼ĻĄ░ņŚÉņä£ ņåīļ│Ćļ¤ē ņ”ØĻ░Ćļź╝ ļ╣äļĪ»ĒĢ£ ņŚ¼ļ¤¼ ļČĆņ×æņÜ®ņØ┤ ļ¦ÄņĢśĻ│Ā, ņØ┤ņŚÉ ļö░ļØ╝ņä£ ņżæļÅä ĒāłļØĮļźĀņØ┤ 23% (ņ£äņĢĮĻĄ░ 13.8%)ņŚÉ ļŗ¼Ē¢łļŗż. ņØ┤ ņĀÉņŚÉ ļīĆĒĢ┤ņä£ ļ»ĖĻĄŁ FDAņŚÉņä£ļŖö Ļ▓░Ļ│╝ ņ¦ĆĒæ£ ņČöņĀĢĻ░ÆņØś ņĀĢĒÖĢņä▒ņŚÉ ņśüĒ¢źņØä ļ»Ėņ│żņØä Ļ░ĆļŖźņä▒ņØä ņÜ░ļĀżĒĢśņśĆļŗż. ĒĢ£ĒÄĖ Ļ░ä ĒÜ©ņåī ņłśņ╣ś ņāüņŖ╣ņØ┤ ņĢĮ 5%ņŚÉņä£ Ļ┤Ćņ░░ļÉśņ¢┤ Ļ│ĀņÜ®ļ¤ē ņןĻĖ░ Ēł¼ņŚ¼ņŚÉ ļö░ļźĖ Ļ░äļÅģņä▒ ņ£äĒŚśņŚÉ ļīĆĒĢ£ ņÜ░ļĀżĻ░Ć ņĀ£ĻĖ░ļÉśņŚłļŗż. Ēł¼ņŚ¼ Ēøä 3-14Ļ░£ņøöņØś ĻĖ░Ļ░äņŚÉ 3Ļ▒┤ņØś ņĢĮļ¼╝ ņ£Āļ░£ņä▒ Ļ░äļÅģņä▒ņØ┤ ĒÖĢņØĖļÉśņŚłņ£╝ļéś, ņżæņ”Ø Ļ░äļČĆņĀäņ£╝ļĪ£ņØś ņ¦äĒ¢ēņØĆ ņŚåņŚłļŗż. ĻĘĖļ¤¼ļéś FDAņŚÉņä£ļŖö 3,000ļ¬ģņŚÉ 1Ļ▒┤ ļ╣äņ£©ļĪ£ ņżæņ”Ø Ļ░äļČĆņĀä ņ£äĒŚśņØä ņśłņāüĒĢśĻ│Ā ņØ┤ņŚÉ ļīĆĒĢ£ ņĀüņĀłĒĢ£ ļīĆņ▓ś(risk evaluation and mitigation strategies)ļź╝ ņÜöĻĄ¼Ē¢łļŗż. Ēśäņ×¼ ļ»ĖĻĄŁņŚÉņä£ļŖö TEMPO ņŚ░ņן ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ē ņżæņØ┤ļ®░, ņ£Āļ¤ĮĻ│╝ ņ║ÉļéśļŗżņŚÉņä£ļŖö ĒؼĻĘĆņĢĮĒÆł ņ¦ĆņĀĢņØä ļ░øņĢśĻ│Ā, ņØ╝ļ│ĖņŚÉņä£ļŖö ĒŚłĻ░ĆņŖ╣ņØĖņØä ļ░øņØĆ ņāüĒā£ļŗż.
ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņ£Āņé¼ņ▓┤(somatostatin analogues)
ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņ£Āņé¼ņ▓┤ņØĖ ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£(octreotide)ļź╝ ļćīĒĢśņłśņ▓┤ ņóģņ¢æ ņ╣śļŻīļź╝ ņ£äĒĢ┤ ņןĻĖ░Ļ░ä Ēł¼ņŚ¼Ē¢łļŹś ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ ļéŁņóģ Ēü¼ĻĖ░ņØś ļ│ĆĒÖöĻ░Ć ņŚåņØīņØä Ļ┤Ćņ░░ĒĢ£ ļÆż ņŚ¼ļ¤¼ ļÅÖļ¼╝ņŗżĒŚśĻ│╝ ņØ╝ļČĆ ņåīĻĘ£ļ¬© ņ×äņāüņŚ░ĻĄ¼ņŚÉņä£ SAĻ░Ć ņĮ®ĒīźĻ│╝ Ļ░äņØś ļéŁņóģņØä ņ¢ĄņĀ£ĒĢ£ļŗżĻ│Ā ļ░ØĒśĆņĪīļŗż[49]. ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņłśņÜ®ņ▓┤ņżæ sst2ļŖö ņĮ®Ēīź ņĪ░ņ¦üĻ│╝ ļŗ┤Ļ┤Ć ļ░Å Ļ░äļéŁņóģ ņäĖĒżņŚÉ ļČäĒżĒĢśĻ│Ā ņ׳ņ£╝ļ®░, ņŚ¼ĻĖ░ņŚÉ ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ņØ┤ Ļ▓░ĒĢ®ĒĢśļ®┤ cAMPļź╝ ņ¢ĄņĀ£ĒĢ£ļŗż. ņØ┤Ēā£ļ”¼ ņŚ░ĻĄ¼ņ×ÉļōżņØ┤ ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£Ļ░Ć ņŗĀļéŁņóģ ņ¢ĄņĀ£ņŚÉ ņ¢┤ļ¢ż ņśüĒ¢źņØä ļ»Ėņ╣śļŖöņ¦Ć ĒÜ©Ļ│╝ļź╝ ļ│┤ĻĖ░ ņ£äĒĢ┤ņä£ 12ļ¬ģņØś ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ 6Ļ░£ņøö ĻĄÉņ░©ņäżĻ│ä ņŚ░ĻĄ¼ļź╝ ņłśĒ¢ēĒ¢łļŗż[49]. ļīĆņāü ĒÖśņ×ÉļōżņØĆ TKV ņĢĮ 2.4 LņŚÉ Ēśłņ▓Ł Ēü¼ļĀłņĢäĒŗ░ļŗī(serum creatinine) 1.9 mg/dLļĪ£ ņ¦äĒ¢ēļÉ£ ļŗ©Ļ│äņśĆļŗż. ļŗ©ĻĖ░Ļ░äņŚÉ ņåīņłśņØś ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ĒīīņØ╝ļ¤┐ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ņ¦Ćļ¦ī, TKV ļ░Å ļéŁņóģ Ēü¼ĻĖ░ņØś ņ”ØĻ░Ćļź╝ ņżäņØ┤ļŖö ĒÜ©Ļ│╝ļź╝ Ļ┤Ćņ░░ĒĢ£ ļ░śļ®┤ ļČĆņ×æņÜ®ņØĆ ņ£äņןĻ┤Ć ņןņĢĀ ņĀĢļÅäļĪ£ Ļ▓Įļ»ĖĒ¢łņ£╝ļ®░ ļīĆļČĆļČä ņĀĆņĀłļĪ£ ĒśĖņĀäļÉśņ¢┤ ņĢłņĀäņä▒ ņĖĪļ®┤ņŚÉņä£ļÅä ņÜ░ņłśĒ¢łļŗż. ņØ┤Ēøä ņØ┤ļōżņØĆ ņןĻĖ░Ļ░äņØś ņ£äņĢĮļīĆņĪ░ņŚ░ĻĄ¼ļź╝ Ļ│äĒÜŹĒ¢łĻ│Ā, ĻĘĖ Ļ▓░Ļ│╝Ļ░Ć A long-acting somatostatin on disease progression in nephropathy due to autosomal dominant polycystic kidney disease ņŚ░ĻĄ¼ņØ┤ļŗż[50]. ņØ┤Ēā£ļ”¼ņØś 5Ļ░£ ļīĆĒĢÖļ│æņøÉņØ┤ ņ░ĖņŚ¼Ē¢łĻ│Ā, MDRD ņČöņĀĢ GFR > 40ņØĖ ņä▒ņØĖņØä ļīĆņāüņ£╝ļĪ£ ļæÉ ĻĄ░ņŚÉ Ļ░üĻ░ü 40, 39ļ¬ģņØś ĒÖśņ×ÉĻ░Ć ļō▒ļĪØļÉśņ¢┤ ņ┤Ø 3ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░Ē¢łļŗż. ņØ╝ņ░© ĒÅēĻ░Ć ņ¦ĆĒæ£ļĪ£ 1ļģä, 3ļģäņ¦Ė TKVņØś ļ│ĆĒÖö ļ░Å Iohexol clearanceļź╝ ņØ┤ņÜ®ĒĢ£ GFRņØś ļ│ĆĒÖöļź╝ ĒÅēĻ░ĆĒ¢łļŗż. 1ļģäĻ│╝ 3ļģäņ¦Ė TKV ļ│ĆĒÖöņ£©ņØä ļ╣äĻĄÉĒĢ┤ ļ│┤ļ®┤, 1ļģäņ¦Ė TKV, ļéŁņóģ ļČĆĒö╝(total cyst volume)ņØś ļ│ĆĒÖö ļ¬©ļæÉ ņ£ĀņØśĒĢśĻ▓ī ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£ĻĄ░ņŚÉņä£ ņ×æņĢśņ£╝ļéś(46.2 vs. 143.7 mL, p = 0.032), 3ļģäņ¦ĖļŖö ņ£ĀņØśņłśņżĆņŚÉ ļÅäļŗ¼ĒĢśņ¦ĆļŖö ļ¬╗Ē¢łļŗż. ņ”ē, ņ▓½ 1ļģäņ¦Ė ņä▒ņןņ¢ĄņĀ£ ĒÜ©Ļ│╝Ļ░Ć Ļ░Ćņן Ēü░ Ļ▓░Ļ│╝ļź╝ ņØśļ»ĖĒĢśĻ│Ā, ņØ┤ļŖö TEMPO ņŚ░ĻĄ¼ņŚÉņä£ļÅä ļ╣äņŖĘĒĢ£ Ļ▓░Ļ│╝ļĪ£ņä£ ņ╣śļŻī ņŗ£ņ×æ ņ┤łĻĖ░ņŚÉ ļéŁņóģ ļé┤ļČĆļĪ£ņØś ņ▓┤ņĢĪ ļČäļ╣äĻ░Ć ĻĖēĻ▓®ĒĢśĻ▓ī ņżäļ®┤ņä£ ļéŁņóģņØ┤ ņ£äņČĢļÉśņ¦Ćļ¦ī, ņŗ£Ļ░äņØ┤ ņ¦ĆļéĀņłśļĪØ ņØ┤ļ¤¼ĒĢ£ ņØ┤ļōØņØ┤ ņżäņ¢┤ļōżĻĖ░ ļĢīļ¼ĖņØ┤ļØ╝Ļ│Ā ļ│Ėļŗż. GFR Ļ▓░Ļ│╝Ļ░Ć ĒØźļ»ĖļĪ£ņÜ┤ļŹ░, 3ļģäĻ╣īņ¦ĆņØś ņĀäņ▓┤ņĀüņØĖ ļ│ĆĒÖöņ£©ņØĆ ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć ņŚåņ¦Ćļ¦ī, ņØ┤ļź╝ ĻĖ░Ļ░äļ│äļĪ£ ļéśļłäņ¢┤ ļČäņäØĒĢ£ Ļ▓░Ļ│╝ ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£ ņ╣śļŻīĻĄ░ņŚÉņä£ 1ļģäĻ╣īņ¦ĆļŖö ņ░©ņØ┤Ļ░Ć ņŚåļŗżĻ░Ć ņØ┤ĒøäņŚÉ ņĢłņĀĢļÉśļŖö ĒśäņāüņØä ļ│╝ ņłś ņ׳ļŗż. 1ļģäņ¦Ė GFRņØś ļ│ĆĒÖöņ£©Ļ│╝ 3ļģäņ¦Ė ļ│ĆĒÖöņ£©ņØś ņāüĻ┤ĆĻ┤ĆĻ│äļź╝ ļČäņäØĒ¢łņØä ļĢī, ņ£äņĢĮĻĄ░ņŚÉņä£ļŖö 1ļģäņ¦Ė GFR Ļ░ÉņåīĻ░Ć Ēü┤ņłśļĪØ 3ļģäņ¦ĖļÅä GFR Ļ░ÉņåīĻ░Ć Ēü░ ļŹ░ ļ░śĒĢ┤ņä£ ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£ĻĄ░ņŚÉņä£ļŖö 1ļģäņ¦Ė GFR Ļ░ÉņåīĻ░Ć Ēü┤ņłśļĪØ 3ļģäņ¦Ė GFR Ļ░ÉņåīĒÅŁņØ┤ ņ×æņØĆ ļ░śļīĆņØś Ļ▓ĮĒ¢źņØä Ļ┤Ćņ░░ĒĢĀ ņłś ņ׳ņŚłļŗż. ņ”ē, ņ╣śļŻī ņ┤łĻĖ░ņŚÉ GFRņØ┤ Ēü░ ĒÅŁņ£╝ļĪ£ Ļ░ÉņåīĒĢĀņłśļĪØ ņןĻĖ░ ĒÜ©Ļ│╝ļź╝ ĻĖ░ļīĆĒĢĀ ņłś ņ׳ļŗżļŖö ļ£╗ņ£╝ļĪ£ ļ¦łņ╣ś ļŗ╣ļć©ņä▒ ņŗĀņ”ØņŚÉņä£ ACEiļź╝ Ēł¼ņŚ¼Ē¢łņØä ļĢīņØś ļ░śņØæĻ│╝ ĒØĪņé¼ĒĢśļŗż. Ēśäņ×¼ ļīĆņāüņ×Éļź╝ 98ļ¬ģĻ╣īņ¦Ć ļŖśļ”░ ĒøäņåŹ ņŚ░ĻĄ¼(NCT 01377246)Ļ░Ć ņŗ£ņ×æļÉśņŚłĻ│Ā, ņØ┤ņÖĖņŚÉļÅä ļśÉ ļŗżļźĖ SAņØĖ ļ×ĆļĀłņśżĒāĆņØ┤ļō£(lanreotide)ļź╝ ņØ┤ņÜ®ĒĢ£ ņ×äņāü ņŚ░ĻĄ¼(DIPAK1, LIPS)Ļ░Ć ņ£Āļ¤ĮņØä ņżæņŗ¼ņ£╝ļĪ£ ņ¦äĒ¢ē ņżæņØ┤ļŗż[51].
SAļŖö mammalian target of rapamycin (mTOR) ņ¢ĄņĀ£ņĀ£ļéś Ēå©ļ▒üĒāä ļō▒ņØś ļŗżļźĖ Ēøäļ│┤ ņĢĮļ¼╝ņŚÉ ļ╣äĒĢ┤ņä£ ņןĻĖ░ ļČĆņ×æņÜ®ņØ┤ ņāüļīĆņĀüņ£╝ļĪ£ ļ»Ėļ»ĖĒĢśļŗżļŖö ņĀÉ ņÖĖņŚÉ ņåīļ¦łĒåĀņŖżĒāĆĒŗ┤ ņłśņÜ®ņ▓┤Ļ░Ć Ļ░äļŗ┤ļÅäņŚÉļÅä ņĪ┤ņ×¼ĒĢśĻ│Ā, ņśźĒŖĖļĀłņśżĒāĆņØ┤ļō£ ļ░Å ļ×ĆļĀłņśżĒāĆņØ┤ļō£(lanreotide)Ļ░Ć Ļ░äļéŁņóģņØś ņä▒ņןņØä ļŖ”ņČöļŖö ļŹ░ļÅä ĒÜ©Ļ│╝Ļ░Ć ņ׳ļŗżļŖö ņןņĀÉņØ┤ ņ׳ļŗż[52,53].
mTOR inhibitor
ņØ┤ņŗØļ®┤ņŚŁņ¢ĄņĀ£ņĀ£ņØĖ ļØ╝Ēīīļ¦łņØ┤ņŗĀ(rapamycin = sirolimus)ņØĆ ņäĖĒż ļé┤ņŚÉņä£ FK506 Ļ▓░ĒĢ® ļŗ©ļ░▒(FK506 binding protein)Ļ│╝ Ļ▓░ĒĢ®ĒĢ£ ļÆż mTORņØä ņ¢ĄņĀ£ĒĢ£ļŗż. mTORļŖö ņäĖĒż ņ”ØņŗØņŚÉ Ļ┤ĆļĀ©ļÉ£ ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņŗĀĒśĖ ĻĖ░ņĀäņØä ņĪ░ņĀłĒĢ£ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░, mTOR ņ¢ĄņĀ£ņĀ£ļŖö ņŗĀņäĖĻ┤Ć ņäĖĒż ņ”ØņŗØņØä ņ¢ĄņĀ£ĒĢśņŚ¼ ņāüņŚ╝ņāēņ▓┤ņÜ░ņä▒ ļŗżļéŁņŗĀņØś ņ╣śļŻīņŚÉ Ēü░ ņŚŁĒĢĀņØä ĒĢĀ Ļ▓āņ£╝ļĪ£ ĻĖ░ļīĆļź╝ ļ¬©ņĢśļŗż. ņŗżņĀ£ļĪ£ mTOR ņ¢ĄņĀ£ņĀ£ļŖö ņäĖĒżņŻ╝ ņŗżĒŚśņØ┤ļéś ļÅÖļ¼╝ ņŗżĒŚśņŚÉņä£ ļéŁņóģ ņāüĒö╝ņäĖĒżņØś ņ”ØņŗØņØä ņ¢ĄņĀ£ĒĢśņŚ¼ ļéŁņóģ ņä▒ņןņØä ņżäņØ┤Ļ│Ā, ņŗĀĻĖ░ļŖźņØś ņĢģĒÖöļź╝ ļ░®ņ¦ĆĒ¢łļŗż[54-56]. ĻĘĖļ¤¼ļéś ņØ┤Ēøä ļ░£Ēæ£ļÉ£ ļæÉ Ļ░Ćņ¦Ć ļīĆĻĘ£ļ¬© ļ¼┤ņ×æņ£ä ņ×äņāüņŗ£ĒŚś Ļ▓░Ļ│╝ļŖö mTOR ņ¢ĄņĀ£ņĀ£ņØś ĒÜ©ļŖźņØä ņ×ģņ”ØĒĢśļŖö ļŹ░ ņŗżĒī©ĒĢśņśĆļŗż. Serra ļō▒[57]ņØ┤ 100ļ¬ģņØś ļ╣äĻĄÉņĀü ņŗĀĻĖ░ļŖźņØ┤ ļ│┤ņĪ┤ļÉśņ¢┤ ņ׳ļŖö(> 70 mL/min/1.73 m2) ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ sirolimusļź╝ 18Ļ░£ņøö Ēł¼ņŚ¼ĒĢśņśĆņØä ļĢī ļīĆņĪ░ĻĄ░ņŚÉ ļ╣äĒĢ┤ ņŗĀņןņÜ®ņĀüņØ┤ļéś ņŗĀĻĖ░ļŖźņŚÉ ņ░©ņØ┤Ļ░Ć ņŚåņŚłļŗż. ļśÉĒĢ£ ĒÅēĻĘĀ GFR 53-56 mL/min/1.73 m2ņØ┤Ļ│Ā TKV 1,900-2,000 mLņŚÉ ĒĢ┤ļŗ╣ĒĢśļŖö ņāüļīĆņĀüņ£╝ļĪ£ ņ¦äĒ¢ēļÉ£ ļŗ©Ļ│äņØś ļŗżļéŁņŗĀ ĒÖśņ×É 433ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ everolimusļź╝ 2ļģäĻ░ä Ēł¼ņŚ¼ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļÅä ņ┤łĻĖ░ 1ļģäņ¦Ė ņŗĀņןņÜ®ņĀüņØś ņ”ØĻ░Ć ņåŹļÅäļź╝ ņżäņØ┤ļŖö ĒÜ©Ļ│╝Ļ░Ć ņ׳ņŚłņ£╝ļéś 2ļģäņ¦ĖņŚÉļŖö ņ░©ņØ┤Ļ░Ć Ļ┤Ćņ░░ļÉśņ¦Ć ņĢŖņĢśĻ│Ā, ņśżĒ׳ļĀż everolimus Ēł¼ņŚ¼ĻĄ░ņŚÉņä£ ņŗĀĻĖ░ļŖź Ļ░Éņåī ņåŹļÅäĻ░Ć ļ╣Āļ”äņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż[58]. Ļ▓īļŗżĻ░Ć ļæÉ ņŚ░ĻĄ¼ ļ¬©ļæÉ mTOR ņ¢ĄņĀ£ņĀ£ļź╝ Ēł¼ņŚ¼ĒĢ£ ĻĄ░ņŚÉņä£ ļČĆņ×æņÜ®ņØ┤ ļŹö ļ¦ÄņĢśĻ│Ā, ņČ®ļČäĒĢ£ ņÜ®ļ¤ēņŚÉ ļÅäļŗ¼ĒĢśļŖö ļŹ░ ņĀ£ĒĢ£ņØ┤ ļÉśĻ│Ā ņ׳ļŗż[59]. ļÅÖļ¼╝ ņŗżĒŚśņŚÉņä£ ĒÜ©ļŖźņØä ļéśĒāĆļéĖ ņÜ®ļ¤ēņØĆ ņØĖĻ░äņŚÉņä£ ņé¼ņÜ®ĒĢ£ ņÜ®ļ¤ēņØś ņĢĮ ņŚ┤ ļ░░ņŚÉ ļŗ¼ĒĢ£ļŗżļŖö ņĀÉņØä Ļ│ĀļĀżĒĢ┤ņĢ╝ ĒĢ£ļŗż[60]. ļČĆņ×æņÜ®ņØä ņżäņØ┤ļ®┤ņä£ ļéŁņóģņäĖĒż ņ¢ĄņĀ£ ĒÜ©Ļ│╝ļź╝ ņĄ£ļīĆĒÖöĒĢśĻĖ░ ņ£äĒĢ┤ņä£ ļØ╝Ēīīļ¦łņØ┤ņŗĀņŚÉ ņŚĮņé░ņØä ņżæĒĢ®ĒĢśņŚ¼ ļéŁņóģ ņäĖĒżļź╝ ĒāĆĻ╣āĒĢśļŖö ņĀäļץņØ┤ ņŗ£ļÅäļÉśĻ│Ā ņ׳ļŗż[61].
Pravastatin
Ļ│ĀĒśłņĢĢņØ┤ ņ׳ļŖö 110ļ¬ģņØś ņåīņĢä ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒöäļØ╝ļ░öņŖżĒāĆĒŗ┤(pravastatin)Ļ│╝ ņ£äņĢĮņØś ĒÜ©Ļ│╝ļź╝ 3ļģäĻ░ä ņČöņĀü ļ╣äĻĄÉĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ ņ╣śļŻīĻĄ░ņŚÉņä£ ņĮ®Ēīź ļČĆĒö╝ņØś ņ”ØĻ░ĆĻ░Ć ņ£ĀņØśĒĢśĻ▓ī ņĀüņŚłļŗż[62]. ĻĘĖļ¤¼ļéś ņä▒ņØĖ ļŗżļéŁņŗĀ ĒÖśņ×É 49ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ ĒĢ£ ļ¼┤ņ×æņ£ä ņŚ░ĻĄ¼ņŚÉņä£ļŖö 2ļģäĻ░äņØś ļ╣äĻĄÉņŚÉņä£ ņ£ĀņØśĒĢ£ ņ░©ņØ┤Ļ░Ć ņŚåņŚłĻĖ░ ļĢīļ¼ĖņŚÉ Ļ▓░ļĪĀņØä ļé┤ĻĖ░ļŖö ņ¢┤ļĀĄļŗż[63]. ņĮ®Ēīź ĻĖ░ļŖźņØ┤ ņĀĢņāüņØĖ ņ┤łĻĖ░ņØś ļŗżļéŁņŗĀ ĒÖśņ×ÉņŚÉņä£ Ļ│Āņ¦ĆĒśłņ”ØņØ┤ ļÅÖļ░śļÉśņ¢┤ ņ׳ļŗżļ®┤, ņ¦Ćņ¦ł ņĪ░ņĀł ņÖĖņŚÉ ļéŁņóģ ņä▒ņן ņ¢ĄņĀ£ļź╝ ņ£äĒĢ┤ņä£ ņŖżĒāĆĒŗ┤ņØä ņĀüĻĘ╣ņĀüņ£╝ļĪ£ Ļ│ĀļĀżĒĢ┤ ļ│╝ ņłś ņ׳Ļ▓Āļŗż.
Ļ▓░ ļĪĀ
ļ│ĖļĪĀņŚÉņä£ ĻĖ░ņłĀĒĢ£ V2RA, SA ņÖĖņŚÉļÅä Src ņ¢ĄņĀ£ņĀ£(bosutinib), KD019 Ļ░ÖņØĆ tyrosine kinase ņ¢ĄņĀ£ņĀ£ļōżņØś ņ×äņāüņŗ£ĒŚśņØ┤ ņ¦äĒ¢ē ņżæņØ┤Ļ│Ā ļŗżņ¢æĒĢ£ Ļ│äņŚ┤ņØś Ēøäļ│┤ ņĢĮļ¼╝ļōżņØ┤ ņĀäņ×äņāü ņŗ£ĒŚś ņżæņŚÉ ņ׳ļŗż. ļ┐Éļ¦ī ņĢäļŗłļØ╝, ĒÜ©ļŖźņØ┤ ĒÖĢņØĖļÉ£ ņä£ļĪ£ ļŗżļźĖ Ļ│äņŚ┤ņØś ņĢĮļ¼╝ņØä ļ│æĒĢ®ĒĢśļ®┤ ņČöĻ░Ć ņØ┤ļōØņØ┤ ņ׳ļŖöņ¦ĆņŚÉ ļīĆĒĢ┤ņä£ ņŗ£ļÅäĻ░Ć ņŗ£ņ×æļÉśĻ│Ā ņ׳ļŗż[64-66].
ļŗżļéŁņŗĀ ĒÖśņ×Éļź╝ ņ¦äļŻīĒĢśĻ│Ā ņ׳ļŖö ņ×äņāüņØśļōżņØĆ ņĀüĻĘ╣ņĀüņ£╝ļĪ£ ĒśłņĢĢņØä ņĪ░ņĀłĒĢśļ®┤ņä£ ļéŁņóģ ņä▒ņןņØä ņ¦üņĀæ ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ļŖö ņāłļĪ£ņÜ┤ ņĢĮļ¼╝ļōżņŚÉ ļīĆĒĢ£ ņ×äņāü ņŗ£ĒŚś ņāüĒÖ®ņØä ņČöņŗ£ĒĢśļ®┤ņä£ ĒÖśņ×ÉļōżņŚÉĻ▓ī ņĄ£ņŗĀ ņĀĢļ│┤ļź╝ ņĀ£Ļ│ĄĒĢ┤ņĢ╝ ĒĢ£ļŗż. ĒŖ╣Ē׳ ņśüņāü Ļ▓Ćņé¼ ņČöņĀü ļ░Å Ļ░ĆņĪ▒ļĀź ņĀĢļ│┤ļź╝ ĒÖ£ņÜ®ĒĢśņŚ¼ Ļ│Āņ£äĒŚśĻĄ░ņØä ņäĀļ│äĒĢśĻ│Ā, ĒĢäņÜöĒĢśļŗżļ®┤ ļŗżļéŁņŗĀ ņĀäļ¼Ė Ēü┤ļ”¼ļŗēņŚÉ ņØśļó░ĒĢśņŚ¼ ĻĄÉņ£Ī ļ░Å Ļ│Āņ£äĒŚśĻĄ░ ļīĆņāüņ×ÉļōżņØ┤ ņ×äņāüņŗ£ĒŚśņŚÉ ņ░ĖņŚ¼ĒĢĀ ņłś ņ׳ļŖö ĻĖ░ĒÜīļź╝ Ļ░¢ļÅäļĪØ ļÅĢļŖö ļō▒ņØś ļ░®ņĢłļÅä Ļ│ĀļĀżĒĢ┤ņĢ╝ ĒĢ£ļŗż. ļ│Ė ņóģņäżņŚÉņä£ ļŗżļŻ©ņ¦Ć ņĢŖņØĆ ļŗżļéŁņŗĀņØś ņ¦äļŗ© ļ░Å ļŗżļźĖ ĒĢ®ļ│æņ”ØņŚÉ ļīĆĒĢ£ Ļ┤Ćļ”¼ņŚÉ ļīĆĒĢ┤ņä£ ņČöĻ░Ć ņĀĢļ│┤Ļ░Ć ĒĢäņÜöĒĢśļŗżļ®┤ 2015ļģä ļ░£Ēæ£ļÉ£ KDIGO ņ¦äļŻī ņ¦Ćņ╣©ņØä ņ░ĖĻ│ĀĒĢśļÅäļĪØ ĻČīĒĢ£ļŗż[2].
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






