


ņä£ ļĪĀ
Helicobacter pylori (H. pylori)ļŖö ļ»ĖņäĖĒśĖĻĖ░ņä▒ ĻĘĖļ×ī ņØīņä▒ ļ¦ēļīĆĻĘĀņ£╝ļĪ£ 1983ļģä ņ▓śņØī ņ£ä ņĀÉļ¦ēņŚÉņä£ ļČäļ”¼, ļ░░ņ¢æļÉ£ ņØ┤Ēøä ļ¦īņä▒ņ£äņŚ╝, ņåīĒÖöņä▒ĻČżņ¢æ, ņ£ä ļ│ĆņŚ░ļČĆ BņäĖĒż ļ”╝Ēöäņóģ, ĻĘĖļ”¼Ļ│Ā ņ£äņĢöņØś ņøÉņØĖņØĖņ×ÉļĪ£ ĻĘ£ļ¬ģļÉśņŚłļŗż[1,2]. H. pyloriļŖö ņĀä ņäĖĻ│ä ņØĖĻĄ¼ņØś ļ░ś ņłś ņØ┤ņāüņØ┤ Ļ░ÉņŚ╝ļÉśņ¢┤ ņ׳ņ£╝ļ®░, ĒĢ£ ļ▓ł Ļ░ÉņŚ╝ļÉśļ®┤ ņłśļģä ļśÉļŖö ņØ╝ņāØ ļÅÖņĢł Ļ░ÉņŚ╝ņØ┤ ņ¦ĆņåŹļÉśĻ│Ā ņ×ÉņŚ░ ņ╣śņ£ĀļÉśļŖö ņØ╝ņØĆ Ļ▒░ņØś ņŚåļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[3]. ņäĀņ¦äĻĄŁņŚÉņä£ļŖö H. pylori Ļ░ÉņŚ╝ļźĀņØ┤ ļé«ņØĆ ļ░śļ®┤ Ļ░£ļ░£ļÅäņāüĻĄŁņØ┤ļéś Ēøäņ¦äĻĄŁņŚÉņä£ļŖö ļåÆņØĆ Ļ░ÉņŚ╝ļźĀņØä ļéśĒāĆļéĖļŗż. ņØ┤ļ¤░ Ļ░ÉņŚ╝ļźĀņØś ņ░©ņØ┤ļŖö Ļ▓ĮņĀ£ ņłśņżĆņØ┤ļéś ņ£äņāØ ļ░Å ĒÖśĻ▓Į ņāüĒā£ņŚÉ ļö░ļØ╝ ņóīņÜ░ļÉśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[4,5]. ĻĘĖļÅÖņĢł ņŚ¼ļ¤¼ ĒøäņåŹ ņŚ░ĻĄ¼ļōżņØä ĒåĄĒĢśņŚ¼ H. pylori Ļ░ÉņŚ╝ņØś ņŚŁĒĢÖņĀü ĒŖ╣ņä▒Ļ│╝ ļ│æĒā£ņāØļ”¼, ĻĘĖļ”¼Ļ│Ā ņ¦äļŗ©Ļ│╝ ņ╣śļŻīļ░®ļ▓Ģ ļō▒ņŚÉņä£ Ļ┤äļ¬®ĒĢĀ ļ¦īĒĢ£ ņ¦äņĀäņØ┤ ņØ┤ļŻ©ņ¢┤ņĪīļŗż. ĒŖ╣Ē׳, ņÜ░ļ”¼ļéśļØ╝ļŖö ņ£äņĢö ļ░£ņāØļźĀņØ┤ ļåÆĻ│Ā, ņĢöņ£╝ļĪ£ ņØĖĒĢ£ ņé¼ļ¦Ø ņøÉņØĖ ņżæ ņ£äņĢöņØ┤ ņ£╝ļ£ĖņØä ņ░©ņ¦ĆĒĢśĻ│Ā ņ׳ņ¢┤ H. pyloriņØś ņ¦äļŗ©Ļ│╝ ņ╣śļŻīĻ░Ć ņ£äņĢö ņśłļ░®ņ░©ņøÉņŚÉņä£ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢśĻ│Ā ņ׳ļŗż. ļ│ĖĻ│ĀņŚÉņä£ļŖö H. pyloriņØś Ļ░ÉņŚ╝ ņŚŁĒĢÖņŚÉ ļīĆĒĢ┤ ĻĄŁļé┤ ņŚ░ĻĄ¼Ļ▓░Ļ│╝ļōżņØä ņżæņŗ¼ņ£╝ļĪ£ ņé┤ĒÄ┤ļ│┤Ļ│Ā, H. pylori Ļ░ÉņŚ╝ņØ┤ ņåīĒÖöņä▒ĻČżņ¢æ ļ░Å ņ£äņĢöņØä ņØ╝ņ£╝ĒéżļŖö ļ│æĒā£ņāØļ”¼ņŚÉ ļīĆĒĢ┤ ņĢīņĢäļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
H. pylori Ļ░ÉņŚ╝ņØś ņŚŁĒĢÖ
H. pyloriņØś Ļ░ÉņŚ╝ļ╣łļÅä
H. pyloriļŖö ĒĢ£ ļ▓ł Ļ░ÉņŚ╝ļÉśļ®┤ ņłśļģä ļśÉļŖö ņØ╝ņāØ ļÅÖņĢł Ļ░ÉņŚ╝ņØ┤ ņ¦ĆņåŹļÉśļ®┤ņä£ ļ¦īņä▒ņ£äņŚ╝, ņåīĒÖöņä▒ ĻČżņ¢æĻ│╝ ņ£äņĢö, ņ£ä ļ”╝ĒöäņóģņØä ņØ╝ņ£╝ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[3]. ĻĘĖļ¤¼ļéś H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉ£ ļīĆļČĆļČäņØś ĒÖśņ×ÉņŚÉņä£ļŖö ņ×äņāüņ”ØņāüņØ┤ ņŚåņ£╝ļ»ĆļĪ£ ņ×äņāüņ”ØņāüņØ┤ ņŚåļŖö ņä▒ņØĖņŚÉņä£ ņ¢╝ļ¦łļéś Ļ░ÉņŚ╝ļÉśņ¢┤ ņ׳ļŖöņ¦ĆĻ░Ć ņżæņÜöĒĢśļŗż. H. pyloriņØś Ļ░ÉņŚ╝ļźĀņØĆ ņØ╝ļ░śņĀüņ£╝ļĪ£ ņŚ░ļĀ╣ņØ┤ ņ”ØĻ░ĆĒĢśļ®┤ņä£ Ļ░ÉņŚ╝ļźĀļÅä ņ”ØĻ░ĆĒĢ£ļŗż. H. pylori Ļ░ÉņŚ╝ņØĆ Ļ░£ļ░£ļÅäņāüĻĄŁņŚÉņä£ļŖö ņŻ╝ļĪ£ ņĢäļÅÖĻĖ░ņŚÉ ļ░£ņāØĒĢśņ¦Ćļ¦ī ņäĀņ¦äĻĄŁņŚÉņä£ļŖö ņĢäļÅÖĻĖ░ņØś Ļ░ÉņŚ╝ņØĆ ņĀüņØĆ ļ░śļ®┤ ņä▒ļģäĻĖ░ņŚÉļÅä Ļ░ÉņŚ╝ņØ┤ ĻŠĖņżĆĒ׳ ļ░£ņāØĒĢ£ļŗż[6,7]. ņØ┤ņÖĆ Ļ░ÖņØĆ Ļ▓░Ļ│╝ļŖö ņä▒ņןĻĖ░ņØś ņé¼ĒÜīĻ▓ĮņĀ£ ņŚ¼Ļ▒┤ ļ░Å ĻĄÉņ£Ī ņłśņżĆĻ│╝ Ļ┤ĆĻ│äĻ░Ć ņ׳ņØä Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż[8-11]. Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ H. pylori Ļ░ÉņŚ╝ ņ£Āļ│æļźĀņØĆ ņäĀņ¦äĻĄŁņØ╝ņłśļĪØ ļé«Ļ│Ā Ļ░£ļ░£ļÅäņāüĻĄŁņŚÉņä£ ļåÆņ£╝ļ®░, ņØ┤ ļ░¢ņŚÉļÅä ņŚ░ļĀ╣Ļ│╝ ņ¦ĆņŚŁņĀü ļČäĒż, ĻĘĖļ”¼Ļ│Ā ņóģņĪ▒ Ļ░äņŚÉ ņ░©ņØ┤ļź╝ ļ│┤ņØĖļŗż[12,13]. H. pyloriļŖö ņØĖņ£äņĀüņ£╝ļĪ£ ņĀ£ĻĘĀņ╣śļŻīļź╝ ĒĢśņ¦Ć ņĢŖļŖö ĒĢ£ ļīĆļČĆļČäņŚÉņä£ ĒÅēņāØļÅÖņĢł Ļ░ÉņŚ╝ņØ┤ ņ¦ĆņåŹļÉśļ»ĆļĪ£ H. pylori Ļ░ÉņŚ╝Ļ│╝ Ļ┤ĆļĀ©ļÉ£ ņØĖņ×ÉļōżņØä ļČäņäØĒĢśļŖö Ļ▓āņØĆ Ļ│Ąņżæļ│┤Ļ▒┤ĒĢÖņĀüņØĖ ņĖĪļ®┤ņŚÉņä£ ņżæņÜöĒĢ£ ņØśļ»ĖĻ░Ć ņ׳ļŗż. ĒĢśņ¦Ćļ¦ī ņ¦Ćļé£ 30ņŚ¼ ļģäĻ░ä H. pyloriņŚÉ Ļ┤ĆĒĢ£ ņŚŁĒĢÖ ļ░Å ļ│æņØĖņŚÉ Ļ┤ĆĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ļ¦ÄņØĆ ņä▒Ļ│╝Ļ░Ć ņ׳ņŚłņ¦Ćļ¦ī Ļ░ÉņŚ╝ Ļ▓ĮļĪ£ ļ░Å ņśłļ░® ņĖĪļ®┤ņŚÉņä£ļŖö ņĢäņ¦ü ļ¬ģĒÖĢĒ׳ ļ░ØĒśĆņ¦ä ļ░öĻ░Ć ņĀüņØĆ ĒÄĖņØ┤ļŗż.
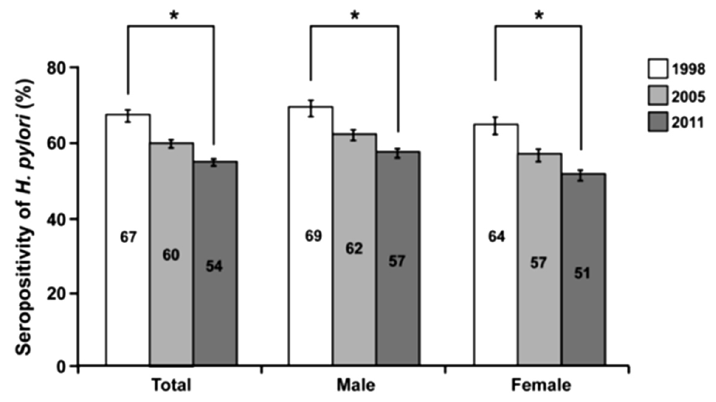
ņÜ░ļ”¼ļéśļØ╝ņŚÉņä£ļŖö 1998ļģä ļīĆĒĢ£ H. pylori ņŚ░ĻĄ¼ĒÜīņŚÉņä£ ņĄ£ņ┤łļĪ£ ņĀäĻĄŁ Ļ░ü ņ¦ĆņŚŁņŚÉņä£ ļ¼┤ņ”ØņāüņØĖ 5,732ļ¬ģņØä ļīĆņāüņ£╝ļĪ£ Ēśłņ▓Ł Ļ▓Ćņé¼ļź╝ ņŗ£Ē¢ēĒĢśņŚ¼ H. pylori Ļ░ÉņŚ╝ļźĀņØä ņĪ░ņé¼ĒĢśņśĆļŗż[12]. ĻĘĖ Ļ▓░Ļ│╝ H. pyloriņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ 46.6%ņśĆņ£╝ļ®░, 15ņäĖ ņØ┤ĒĢśņØś ņåīņĢä(2,338ļ¬ģ)ņŚÉņä£ļŖö 17.2%, 16ņäĖ ņØ┤ņāüņØś ņä▒ņØĖ(3,394ļ¬ģ)ņŚÉņä£ļŖö 66.9%ļź╝ ļéśĒāĆļāłļŗż. 2005ļģä ņĀäĻĄŁņĀüņ£╝ļĪ£ 15,916ļ¬ģņØś Ļ▒┤Ļ░ĢĻ▓Ćņ¦äņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ņŗ£Ē¢ēĒĢ£ ņŚ░ĻĄ¼[14]ņŚÉņä£ļŖö 16ņäĖ ņØ┤ņāü ņĀ£ĻĘĀņ╣śļŻīņØś Ļ│╝Ļ▒░ļĀźņØ┤ ņŚåļŖö ļ¼┤ņ”Øņāü ņä▒ņØĖņØś H. pylori Ēśłņ▓Ł ņ£Āļ│æļźĀņØĆ 59.6%ļĪ£ 1998ļģäņŚÉ ļ╣äĒĢ┤ Ļ░ÉņåīĒĢśņśĆņ£╝ļ®░, 2011ļģä 10,796ļ¬ģņØś ļ¼┤ņ”Øņāü Ļ▒┤Ļ░ĢĻ▓Ćņ¦ä ņłśņ¦äņ×ÉņØś H. pylori Ēśłņ▓Ł ņ£Āļ│æļźĀņØĆ 54.4%ļĪ£ 1998ļģä ļ░Å 2005ļģäĻ│╝ ļ╣äĻĄÉĒĢĀ ļĢī ņ£ĀņØśĒĢ£ Ļ░Éņåīļź╝ ļ│┤ņśĆļŗż(p < 0.05) [15]. ļśÉĒĢ£, ņŚ░ļĀ╣ļ│ä ņ£Āļ│æļźĀ ņĪ░ņé¼ņŚÉņä£ļÅä ļ¬©ļōĀ ņŚ░ļĀ╣ņŚÉņä£ Ļ░ÉņŚ╝ļźĀņØ┤ Ļ░ÉņåīĒĢśļŖö ņČöņäĖļź╝ ļ│┤ņśĆņ£╝ļ®░, ĒŖ╣Ē׳ 40ņäĖ ļ»Ėļ¦īņØś ņŚ░ļĀ╣ņŚÉņä£ Ēü░ ĒÅŁņ£╝ļĪ£ Ļ░ÉņåīĒĢśņśĆļŗż(Fig. 1). ņØ┤ļ¤¼ĒĢ£ Ļ░Éņåī ņČöņäĖļŖö ĻĘĖ ļÅÖņĢł Ļ▓ĮņĀ£ ņłśņżĆņØ┤ Ē¢źņāüļÉśĻ│Ā ņØ┤ņŚÉ ļö░ļźĖ ņ£äņāØ ņāüĒā£Ļ░Ć ĒśĖņĀäļÉśņŚłĻĖ░ ļĢīļ¼Ėņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż.
H. pyloriņØś Ļ░ÉņŚ╝Ļ▓ĮļĪ£
H. pylori Ļ░ÉņŚ╝ņØś ņĀäĒīī Ļ▓ĮļĪ£ņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ņŚÉļŖö ļ¬ć Ļ░Ćņ¦Ć ļ¼ĖņĀ£ņĀÉņØ┤ ņ׳ļŖöļŹ░ ļ¼┤ņŚćļ│┤ļŗż H. pylori Ļ░ÉņŚ╝ņØĆ ļīĆļČĆļČä ļ¼┤ņ”ØņāüņØ┤ņ¢┤ņä£ ņ┤łĻĖ░ Ļ░ÉņŚ╝ļÉ£ ĒÖśņ×Éļź╝ ņ░ŠņĢäļé┤ĻĖ░Ļ░Ć ņ¢┤ļĀĄļŗżļŖö Ļ▓āņØ┤ļŗż. ļīĆļČĆļČäņØś Ļ░ÉņŚ╝ņØĆ ņĢäļÅÖĻĖ░ņŚÉ ņØ╝ņ¢┤ļéśņ¦Ćļ¦ī, ņåīņĢäļŖö ņä▒ņØĖļ│┤ļŗż ņ¦äļŗ©ņØś ņĀĢĒÖĢņä▒ņØ┤ ļ¢©ņ¢┤ņ¦ĆĻĖ░ ļĢīļ¼ĖņŚÉ ņĢäļÅÖĻĖ░ņŚÉņä£ļŖö H. pylori Ļ░ÉņŚ╝ņØś ņ¦äļŗ©ņØ┤ ņēĮņ¦Ć ņĢŖļŗż. ļśÉ ļŗżļźĖ ļ¼ĖņĀ£ņĀÉņØĆ ļŗżļźĖ Ļ░ÉņŚ╝ņä▒ ņ¦łĒÖśĻ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ H. pyloriņŚÉ Ļ░ÉņŚ╝ņØ┤ ņØ╝ņ¢┤ļéśĻĖ░ ņ£äĒĢ£ ņĪ░Ļ▒┤ļōż, ņ”ē H. pyloriņŚÉ ļģĖņČ£ļÉśļŖö ļ╣łļÅäņÖĆ Ļ░ÉņŚ╝ ņĀĢļÅä, ņłÖņŻ╝ņØś ņĀĆĒĢŁņä▒ ļ░Å H. pyloriĻ░Ć ņ£äņŚÉņä£ ņé┤ņĢäļé©ņĢä ņ”ØņŗØĒĢśļŖö ļŹ░ ĒĢäņÜöĒĢ£ ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņÜöņåīļōżņØ┤ ļ¬ģĒÖĢĒ׳ ĻĘ£ļ¬ģņØ┤ ļÉśņ¢┤ ņ׳ņ¦Ć ņĢŖļŗż. ĻĘĖļ¤¼ļéś Ēśäņ×¼Ļ╣īņ¦Ć ļ░ØņŚ¼ņ¦ä H. pyloriņØś Ļ░ÉņŚ╝ Ļ▓ĮļĪ£ļŖö ņ▓½ņ¦Ė, H. pyloriĻ░Ć ņé¼ļ×īņŚÉņä£ ņé¼ļ×īņ£╝ļĪ£ ņĀäņŚ╝ļÉśļ®░, ļæśņ¦Ė, ņĢäļÅÖĻĖ░ņŚÉ ņŻ╝ļĪ£ Ļ░ĆņĪ▒ ļé┤ņŚÉņä£ Ļ░ÉņŚ╝ņØ┤ ņØ╝ņ¢┤ļé£ļŗżļŖö ņé¼ņŗżņØĆ ņל ņĢīļĀżņĀĖ ņ׳ļŗż.
ņé¼ļ×īĻ│╝ ņé¼ļ×ī ņé¼ņØ┤ņŚÉņä£ H. pyloriĻ░Ć ņĀäņŚ╝ļÉśļŖö Ļ▓ĮļĪ£ļŖö ĒĢŁļ¼Ė-ĻĄ¼Ļ░Ģ Ļ▓ĮļĪ£ņÖĆ ĻĄ¼Ļ░Ģ-ĻĄ¼Ļ░Ģ Ļ▓ĮļĪ£Ļ░Ć ņ׳ļŗż. ĒĢŁļ¼Ė-ĻĄ¼Ļ░Ģ ņĀäņŚ╝ņØĆ ļīĆļ│Ćņ£╝ļĪ£ ļ░░ņČ£ļÉ£ H. pyloriĻ░Ć ņé¼ļ×īļōżņØś ņ¦üņĀæņĀüņØĖ ņĀæņ┤ēņØ┤ļéś ļ¼╝ ļśÉļŖö ņØīņŗØļ¼╝ Ļ░ÖņØĆ ļŗżļźĖ ļ¦żĻ░£ņ▓┤ļź╝ ĒåĄĒĢśņŚ¼ ļŗżļźĖ ņé¼ļ×īņØś ņ£äņŚÉ Ļ░ÉņŚ╝ņØä ņØ╝ņ£╝Ēé©ļŗż[16-18]. ĻĄ¼Ļ░Ģ-ĻĄ¼Ļ░Ģ ņĀäņŚ╝ņØĆ ņĢäņØ┤ņŚÉĻ▓ī ļ»Ėļ”¼ ņö╣ņØĆ ņØīņŗØņØä ļ©╣ņØ┤ļŖö ļÅÖņĢł ĻĘĀņØ┤ ņĀäņŚ╝ļÉĀ ņłś ņ׳ļŗżļŖö Ļ▓āņØ┤ļŗż. ĻĘĖļ¤¼ļéś ņŗżņĀ£ļĪ£ H. pylori Ļ░ÉņŚ╝ņØ┤ ņ׳ļŖö ņé¼ļ×īņØś ņ╣©ņØ┤ļéś ņ╣śņäØņŚÉņä£ H. pyloriĻ░Ć Ļ▓ĆņČ£ļÉśļŖö ļ╣äņ£©ņØĆ ĻĘ╣Ē׳ ļé«ņĢäņä£ ņŗżņĀ£ Ļ░ÉņŚ╝ļ╣łļÅäļŖö ļé«ņØä Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż[19]. ĒØźļ»ĖļĪŁĻ▓īļÅä H. pylori Ļ░ÉņŚ╝ņØ┤ ĻĄ¼Ļ░ĢņŚÉņä£ ĻĄ¼Ļ░Ģņ£╝ļĪ£ ņĀäĒīīļÉ£ļŗżļ®┤ ļé┤ņŗ£Ļ▓Į Ļ▓Ćņé¼ļź╝ ĒåĄĒĢ£ Ļ░ÉņŚ╝ņØä ņČöņĖĪĒĢ┤ ļ│╝ ņłś ņ׳ļŗż. ļé┤ņŗ£Ļ▓Į ĻĖ░ĻĖ░ņŚÉ ņØśĒĢ£ ņĀäĒīīļŖö ļé┤ņŗ£Ļ▓Į Ļ▓Ćņé¼ Ēøä ļČłņČ®ļČäĒĢ£ ņåīļÅģ, ļé┤ņŗ£Ļ▓Į ņäĖņ▓ÖĻĖ░ņØś ņśżņŚ╝ ļśÉļŖö ļé┤ņŗ£Ļ▓Į ĻĖ░ĻĖ░ņØś ļ│Ąņ×ĪĒĢ£ ĻĄ¼ņĪ░ļĪ£ ņØĖĒĢśņŚ¼ ņČ®ļČäĒĢ£ ņåīļÅģņØ┤ ļÉśņ¦Ć ņĢŖņĢä ļ░£ņāØĒĢĀ ņłś ņ׳ņ¦Ćļ¦ī ļé┤ņŗ£Ļ▓Į ĻĖ░ĻĖ░ ņåīļÅģ ņ¦Ćņ╣©ņØä ņČ®ņŗżĒ׳ ņżĆņłśĒĢśļ®┤ H. pyloriļŖö ņČ®ļČäĒ׳ ļ░Ģļ®ĖĒĢĀ ņłś ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[20]. ĒĢ£ĒÄĖ, H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉ£ ņé¼ļ×īņØĆ Ļ░ÉņŚ╝ļÉśņ¦Ć ņĢŖņØĆ ņé¼ļ×īņŚÉ ļ╣äĒĢ┤ ļ░░ņÜ░ņ×Éļéś ņ×ÉļģĆņØś Ļ░ÉņŚ╝ļźĀņØ┤ ļåÆļŗżļŖö ņĀÉņØĆ H. pylori Ļ░ÉņŚ╝ņØ┤ Ļ░ĆņĪ▒ ļé┤ņŚÉņä£ ņØ┤ļŻ©ņ¢┤ņ¦äļŗżļŖö ņé¼ņŗżņØä ļÆĘļ░øņ╣©ĒĢśļŖö ņ”ØĻ▒░ļØ╝Ļ│Ā ĒĢĀ ņłś ņ׳ļŗż[21,22].
H. pylori Ļ░ÉņŚ╝ņŚÉ ņśüĒ¢źņØä ļ»Ėņ╣śļŖö ņØĖņ×É
ņåīņĢäĻĖ░ņØś ĒÖśĻ▓Į
H. pylori Ļ░ÉņŚ╝ņØĆ ņĢäļÅÖĻĖ░ņØś ĒÖśĻ▓ĮņĀüņØĖ ņÜöņåī, ņ”ē ļ░Ćņ¦æļÉ£ Ļ▒░ņŻ╝ ĒÖśĻ▓ĮņØ┤ļéś ņØĖĻĄ¼ ļ░ĆļÅäņÖĆ Ļ┤ĆĻ│äĻ░Ć ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[9]. ņĢäļÅÖĻĖ░ņØś ļé«ņØĆ ņé¼ĒÜīĻ▓ĮņĀ£ņĀü ņłśņżĆĻ│╝ ļ░Ćņ¦æļÉ£ Ļ▒░ņŻ╝ ĒÖśĻ▓ĮņØ┤ ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢĀ Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉśļ®░ ņäĀņ¦äĻĄŁņŚÉņä£ ņŚ░ļĀ╣ļ│ä Ļ░ÉņŚ╝ļźĀņØ┤ ļé«ņØĆ Ļ▓āņØĆ ņĢäļÅÖĻĖ░ņØś Ē¢źņāüļÉ£ Ļ▒░ņŻ╝ ĒÖśĻ▓ĮņØś ļ░śņśüņØ┤ļØ╝Ļ│Ā ļ│╝ ņłś ņ׳ļŗż.
ņØīņŻ╝ņÖĆ ĒØĪņŚ░
ņØīņŻ╝ņŚÉ Ļ┤ĆĒĢśņŚ¼ ņĢīņĮöņś¼ ņäŁņĘ©ņÖĆ H. pylori Ļ░ÉņŚ╝Ļ│╝ņØś Ļ┤ĆĻ│äļŖö ļ¼┤Ļ┤ĆĒĢśļŗżļŖö Ļ▓¼ĒĢ┤ņÖĆ ĒĢŁņäĖĻĘĀ ĒÜ©Ļ│╝ņŚÉ ņØśĒĢ┤ H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ┤ ļ░®ņ¢┤ ņŚŁĒĢĀņØä ĒĢ£ļŗżļŖö ļ│┤Ļ│ĀļÅä ņ׳ņ¢┤ ņĢäņ¦ü ļ¬ģĒÖĢĒĢśņ¦Ć ņĢŖļŗż[23]. ĒØĪņŚ░Ļ│╝ H. pylori Ļ░ÉņŚ╝Ļ│╝ņØś Ļ┤ĆļĀ©ņä▒ņØĆ ņĢäņ¦ü ļģ╝ļ×ĆņØ┤ ņ׳ļŗż[23].
ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£
ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£ņØś ņé¼ņÜ®ņØ┤ H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ Ļ░Éņłśņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżĻ▒░ļéś ņĀÉļ¦ē ņåÉņāüņØä ņĢģĒÖöņŗ£Ēéżņ¦Ć ņĢŖļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[24]. ņĄ£ĻĘ╝ ņןĻĖ░Ļ░ä ļ╣äņŖżĒģīļĪ£ņØ┤ļō£ņä▒ ņåīņŚ╝ņĀ£ņØś ņé¼ņÜ®ņ×ÉņŚÉĻ▓ī H. pylori ļ░Ģļ®Ė ņÜöļ▓Ģņ£╝ļĪ£ ĻČżņ¢æņØś ļ░£ņāØļźĀņØä ļé«ņČśļŗżļŖö ļ│┤Ļ│ĀĻ░Ć ņ׳ļŗż[25].
ņÜ░ļ”¼ļéśļØ╝ H. pylori Ļ░ÉņŚ╝ņØś ņŚ░ļĀ╣ļ│ä ĒŖ╣ņ¦Ģ
1998ļģä ļīĆĒĢ£ H. pylori ņŚ░ĻĄ¼ĒÜīļŖö Ēśłņ▓ŁĒĢÖņĀü ļ░®ļ▓Ģņ£╝ļĪ£ ņĀä ņŚ░ļĀ╣ņĖĄņØä ĒżĒĢ©ĒĢśļŖö ņĀäĻĄŁ ĻĘ£ļ¬©ņØś H. pylori ņŚŁĒĢÖ ņŚ░ĻĄ¼ļź╝ ņłśĒ¢ēĒĢśņśĆļŗż[12]. ĻĘĖ Ļ▓░Ļ│╝ ņĀäņ▓┤ņĀüņ£╝ļĪ£ļŖö ņŚ░ļĀ╣ņØ┤ ņ”ØĻ░ĆĒĢ©ņŚÉ ļö░ļØ╝ H. pyloriņŚÉ ļīĆĒĢ£ ĒĢŁņ▓┤ ņ¢æņä▒ļźĀņØ┤ ņ”ØĻ░ĆĒĢśļŗżĻ░Ć 40ļīĆ ņØ┤ĒøäļĪ£ļŖö Ļ░ÉņåīĒĢśļŖö ņ¢æņāüņØä ļ│┤ņśĆļŗż. ņČ£ņāØ ņ¦üĒøäļČĆĒä░ 6Ļ░£ņøöĻ╣īņ¦ĆļŖö 24.4%, 7-11Ļ░£ņøö 7.4%, 1-3ņäĖ 6%, 4-6ņäĖ 11.5%, 7-9ņäĖ 12.7%, 10-12ņäĖ 27.3%, 13-15ņäĖ 31.1%, 16-19ņäĖ 45.8%, 20ļīĆ 54.3%, 30ļīĆ 74%, 40ļīĆ 78.5%, 50ļīĆ 73.9%, 60ļīĆ 71.7%, 70ļīĆ 67%ļĪ£ ļéśĒāĆļéś 40ļīĆņŚÉņä£ 78.5%ļĪ£ Ļ░Ćņן ļåÆņØĆ ņ¢æņä▒ļźĀņØä ļ│┤ņśĆļŗż. ņČ£ņāØ ņ¦üĒøäņØś ļåÆņØĆ ņ¢æņä▒ļźĀņØĆ ļ¬©ņ▓┤ļĪ£ļČĆĒä░ ņĀäļŗ¼ļÉ£ ĒĢŁņ▓┤Ļ░Ć Ļ▓ĆņČ£ļÉ£ Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż. ļ¬©ņ▓┤ņØś ĒĢŁņ▓┤Ļ░Ć ņ¢┤ļŖÉ ņŗ£ĻĖ░Ļ╣īņ¦Ć ņ¦ĆņåŹļÉśļŖöņ¦ĆļŖö ĒÖĢņŗżĒ׳ ļ░ØĒśĆņ¦Ćņ¦Ć ņĢŖņĢśņ£╝ļéś ņØ╝ļ░śņĀüņ£╝ļĪ£ ņČ£ņāØ Ēøä 4Ļ░£ņøöĻ░ä ņ¦ĆņåŹļÉśļŗżĻ░Ć ĻĘĖ ņØ┤ĒøäņŚÉ Ļ░ÉņåīĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ļśÉ ļŗżļźĖ ĒŖ╣ņ¦ĢņØĆ H. pylori Ļ░ÉņŚ╝ņØ┤ ņŚ░ļĀ╣ņŚÉ ļö░ļØ╝ ņ”ØĻ░ĆĒĢśņŚ¼ 40ļīĆņŚÉņä£ 80% Ļ░ĆĻ╣īņØ┤ ļåÆņØĆ Ļ░ÉņŚ╝ļźĀņØä ļ│┤ņØ┤ļŗżĻ░Ć ĻĘĖ ņØ┤Ēøä ņĀÉņ░© Ļ░ÉņåīļÉśļŖö ĒśäņāüņØ┤ļŗż. ņØ┤ņÖĆ Ļ░ÖņØ┤ Ļ░ÉņŚ╝ļźĀņØ┤ Ļ░ÉņåīĒĢśļŖö ņØ┤ņ£ĀļŖö Ļ░ü ņŚ░ļĀ╣ņĖĄņŚÉņä£ Ēā£ņ¢┤ļé£ ņŗ£ĻĖ░ņØś ĒÖśĻ▓Į ļ│ĆĒÖöņŚÉ ņØśĒĢ£ Ļ▓āņØ┤ļØ╝ĻĖ░ļ│┤ļŗżļŖö ļéśņØ┤Ļ░Ć ļōżņ¢┤Ļ░ÉņŚÉ ļö░ļØ╝ ņ£ä ņĀÉļ¦ēņØś ņ£äņČĢņØ┤ ņ”ØĻ░ĆĒĢśņŚ¼ H. pyloriņØś ņä£ņŗØ ĒÖśĻ▓ĮņØś ļ│ĆĒÖöļź╝ ņ┤łļלĒĢśņśĆĻĖ░ ļĢīļ¼Ėņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż. 2005ļģäņØś ĻĄŁļé┤ ļŗżĻĖ░Ļ┤Ć Ļ│ĄļÅÖ ņŚ░ĻĄ¼[14]ņŚÉņä£ ņŚ░ļĀ╣ļ│ä H. pyloriņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ 16-19ņäĖ 12.5%, 20ļīĆ 26.3%, 30ļīĆ 49.4%, 40ļīĆ 61.3%, 50ļīĆ 65.6%, 60ļīĆ 64.8%, 70ļīĆ 59.7%ņśĆļŗż. ņØ┤Ēøä 2011ļģä 16ņäĖ ņØ┤ņāüņØś ļ¼┤ņ”Øņāü ņłśĻ▓Ćņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ļŗżĻĖ░Ļ┤Ć Ļ│ĄļÅÖ ņŚ░ĻĄ¼[15]ņŚÉņä£ļÅä ņŚ░ļĀ╣ļ│ä H. pyloriņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ 16-19ņäĖ 11.8%, 20ļīĆ 26.4%, 30ļīĆ 42.1%, 40ļīĆ 52.6%, 50ļīĆ 61.4%, 60ļīĆ 61.6%, 70ļīĆ 58.6%ņśĆļŗż. 1998ļģä, 2005ļģä, 2011ļģäņŚÉņä£ņØś ņŚ░ļĀ╣ļ│ä, ņä▒ļ│ä H. pyloriņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØä ļ╣äĻĄÉĒĢ┤ļ│┤ļ®┤ birth cohortĒÜ©Ļ│╝ļź╝ Ļ░ÉņĢłĒĢśĻ│Āņä£ļØ╝ļÅä ņĀä ņŚ░ļĀ╣ņĖĄņŚÉņä£ ļ¬©ļæÉ ĒĢŁņ▓┤ ņ¢æņä▒ļźĀņØ┤ ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīĒĢśņśĆļŗż(p < 0.05, Fig. 1).
ņÜ░ļ”¼ļéśļØ╝ņØś H. pylori Ļ░ÉņŚ╝ņØś ņ¦Ćļ”¼ņĀüņØĖ ļČäĒż
1998ļģä ļīĆĒĢ£ H. pylori ņŚ░ĻĄ¼ĒÜīĻ░Ć ņŗ£Ē¢ēĒĢ£ ņĀäĻĄŁņĀüņØĖ ņŚŁĒĢÖ ņŚ░ĻĄ¼[12]ņŚÉņä£ ņ¦ĆņŚŁņŚÉ ļö░ļźĖ H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ ņä£ņÜĖ 41.9%, Ļ▓ĮĻĖ░ļÅä 47.3%, Ļ░ĢņøÉļÅä 53.4%, ņČ®ņ▓ŁļÅä 43.7%, Ļ▓ĮņāüļÅä 47.1%, ņĀäļØ╝ļÅä 50.6%, ņĀ£ņŻ╝ļÅä 52.9%ļź╝ ļéśĒāĆļé┤ ņ¦ĆņŚŁ Ļ░ä Ļ░ÉņŚ╝ļźĀņŚÉ ņ░©ņØ┤ļź╝ ļ│┤ņśĆļŗż. Ļ░ÉņŚ╝ļźĀņØĆ Ļ░ĢņøÉļÅä, ņĀ£ņŻ╝ļÅä, ņĀäļØ╝ļÅä ņł£ņ£╝ļĪ£ ļåÆņĢśĻ│Ā ņä£ņÜĖņØ┤ Ļ░Ćņן ļé«ņĢśļŗż. 2005ļģä ņŗ£Ē¢ēļÉ£ ĻĄŁļé┤ ļŗżĻĖ░Ļ┤Ć Ļ│ĄļÅÖņŚ░ĻĄ¼[14]ņŚÉņä£ H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ ņä£ņÜĖ 57.6%, Ļ▓ĮĻĖ░ļÅä 62.1%, Ļ░ĢņøÉļÅä 60.8%, ņČ®ņ▓ŁļÅä 65.4%, Ļ▓ĮņāüļÅä 67.7%, ņĀäļØ╝ļÅä 73.6%, ņĀ£ņŻ╝ļÅä 66.9%ļź╝ ļéśĒāĆļé┤ņ¢┤ ņ¦ĆņŚŁ Ļ░ä ņ░©ņØ┤ļź╝ ļ│┤ņśĆļŗż. 2011ļģä ņŗ£Ē¢ēļÉ£ ĻĄŁļé┤ ļŗżĻĖ░Ļ┤Ć Ļ│ĄļÅÖņŚ░ĻĄ¼[15]ņŚÉņä£ļÅä H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ ņä£ņÜĖ 50.0%, Ļ▓ĮĻĖ░ļÅä 60.4%, Ļ░ĢņøÉļÅä 53.4%, ņČ®ņ▓ŁļÅä 55.4%, Ļ▓ĮņāüļÅä 65.1%, ņĀäļØ╝ļÅä 66.2%, ņĀ£ņŻ╝ļÅä 58.9%ļź╝ ļéśĒāĆļé┤ņ¢┤ ņŚ¼ņĀäĒ׳ ņ¦ĆņŚŁ Ļ░ä ņ░©ņØ┤ļź╝ ļéśĒāĆļāłļŗż. 1998ļģä, 2005ļģä, 2011ļģä ņŚ░ĻĄ¼ņØś ņ¦ĆņŚŁļ│ä H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ¢æņä▒ļźĀņØĆ Ļ▓ĮņāüļÅäņÖĆ Ļ░ĢņøÉļÅäļź╝ ņĀ£ņÖĖĒĢśĻ│Ā ņ£ĀņØśĒĢśĻ▓ī Ļ░ÉņåīĒĢśļŖö Ļ▓ĮĒ¢źņØä ļéśĒāĆļāłļŗż(Fig. 2). H. pyloriņØś Ļ░ÉņŚ╝ļźĀņØĆ ņé¼ĒÜīĻ▓ĮņĀ£ ņłśņżĆĻ│╝ ļ░ĆņĀæĒĢ£ Ļ┤ĆļĀ©ņØ┤ ņ׳ņ£╝ļ»ĆļĪ£ ņ¦ĆņŚŁ Ļ░ä ņ░©ņØ┤ļŖö ņä£ņÜĖ ņ¦ĆņŚŁĻ│╝ ņ¦Ćļ░® Ļ░ä ņé¼ĒÜīĻ▓ĮņĀ£ņĀü ņŚ¼Ļ▒┤ņØś ņ░©ņØ┤ ļĢīļ¼ĖņØ╝ Ļ▓āņ£╝ļĪ£ ņČöņĖĪļÉ£ļŗż.
Ē¢źĒøä ņÜ░ļ”¼ļéśļØ╝ņØś H. pylori Ļ░ÉņŚ╝ļźĀņŚÉ ļīĆĒĢ£ ļ│ĆļÅÖ ņČöņØ┤
ņĄ£ĻĘ╝ ņāØĒÖ£ ĒÖśĻ▓Į, ņ”ē ņ£äņāØ ņāüĒā£ņØś Ļ░£ņäĀņ£╝ļĪ£ H. pylori Ļ░ÉņŚ╝ļźĀņØ┤ Ļ░ÉņåīĒĢśļŖö ņČöņäĖņØ┤ļŗż. ļśÉĒĢ£ H. pylori Ļ░ÉņŚ╝ņŚÉ ņØśĒĢ£ ņåīĒÖöņä▒ ĻČżņ¢æ ļ░Å ņ£äņĢö ļ░£ņāØ ļō▒ ņåīĒÖöĻĖ░ņ¦łĒÖśĻ│╝ņØś Ļ┤ĆļĀ©ņä▒ņØ┤ ļäÉļ”¼ ņĢīļĀżņ¦Ćļ®┤ņä£ ļ│┤ļŗż ņĀüĻĘ╣ņĀüņØĖ ņĀ£ĻĘĀņ╣śļŻīļź╝ ĒĢśĻ│Ā ņ׳ļŖö ņŗżņĀĢņØ┤ļŗż. ņØ┤ņŚÉ ļö░ļØ╝ ņĄ£ĻĘ╝ ņÜ░ļ”¼ļéśļØ╝ņŚÉņä£ H. pylori Ļ░ÉņŚ╝ņØ┤ ņĀÉņ░© Ļ░ÉņåīĒĢśļŖö Ļ▓ĮĒ¢źņØä ļ│┤ņØ┤Ļ│Ā ņ׳ļŗż. ĻĘĖļ¤¼ļéś Ē¢źĒøä H. pylori Ļ░ÉņŚ╝ļźĀņØ┤ ņ¢┤ļ¢╗Ļ▓ī ļ│ĆĒĢĀ Ļ▓āņØĖĻ░ĆļŖö ĒĢśļŖö ļ¼ĖņĀ£ļŖö ņĀ£ĻĘĀņ╣śļŻīņØś ņä▒ņĀüņØ┤ Ļ░ÉņåīļÉśĻ│Ā ņ׳ļŖö ĒśäņŗżņØä Ļ│ĀļĀżĒĢĀ ļĢī ņēĮĻ▓ī ĒīÉļŗ©ĒĢĀ ņłś ņ׳ļŖö ļ¼ĖņĀ£Ļ░Ć ņĢäļŗłļ»ĆļĪ£ ņĢ×ņ£╝ļĪ£ļÅä ļ│ĆĒÖö ņČöņØ┤ļź╝ ņśłņØśņŻ╝ņŗ£ĒĢĀ ĒĢäņÜöĻ░Ć ņ׳ļŗż.
H. pylori Ļ░ÉņŚ╝ņØś ļ│æĒā£ ņāØļ”¼
H. pyloriļŖö ņĀä ņäĖĻ│ä ņØĖĻĄ¼ņØś 50% ņØ┤ņāüņØ┤ Ļ░ÉņŚ╝ļÉśņ¢┤ ņ׳ņ¦Ćļ¦ī Ļ░ÉņŚ╝ņŚÉ ņØśĒĢ£ ņ×äņāü ņ¢æņāüņØĆ ļ¼┤ņ”Øņāü ņ£äņŚ╝ņŚÉņä£ ņ£äĻČżņ¢æ, ņ£äņĢöĻ╣īņ¦Ć ļŗżņ¢æĒĢśĻ▓ī ļéśĒāĆļé£ļŗż. ņØ┤ņ▓śļ¤╝ H. pylori Ļ░ÉņŚ╝ Ēøä ļŗżņ¢æĒĢ£ ņ×äņāü ņ¢æņāüņØä ļ│┤ņØ┤ļŖö Ļ▓āņØĆ ņ¦łļ│æ ļ░£ņāØņŚÉ ņ׳ņ¢┤ ņäĖĻĘĀĻ│╝ ņłÖņŻ╝ ņé¼ņØ┤ņŚÉ ņśüĒ¢źņØä ņŻ╝ļŖö ņŚ¼ļ¤¼ ņØĖņ×ÉĻ░Ć Ļ┤ĆņŚ¼ĒĢśĻ│Ā ņ׳ņØīņØä ņØśļ»ĖĒĢ£ļŗż[26]. ņäĖĻĘĀņØś ļÅģņä▒ņØĖņ×ÉĻ░Ć ņ¦łļ│æ ļ░£ņāØņŚÉ ņżæņÜöĒĢ£ ņØ┤ņ£ĀļŖö H. pylori ņ£ĀņĀäņ▓┤ņŚÉ ņ£ĀņĀäņĀü ļŗżĒśĢņä▒(genetic polymorphism)ņØ┤ ņĪ┤ņ×¼ĒĢśĻĖ░ ļĢīļ¼ĖņØ┤ļŗż. ļśÉĒĢ£ ļÅģņä▒ņØĖņ×É ņ¢æņä▒ņØĖ H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉśļŹöļØ╝ļÅä ļīĆļČĆļČä ņ¦łļ│æņØ┤ ļ░£ņāØĒĢśņ¦Ć ņĢŖļŖö Ļ▓āņØĆ ņłÖņŻ╝ņØĖņ×ÉņŚÉ ļö░ļØ╝ ļ░£ļ│æņŚÉ ņ░©ņØ┤Ļ░Ć ņ׳ĻĖ░ ļĢīļ¼ĖņØ┤ļŗż. ņŚ¼ĻĖ░ņä£ļŖö H. pyloriņØś ņäĖĻĘĀĒĢÖņĀü ĒŖ╣ņä▒Ļ│╝ ņłÖņŻ╝ņØś ļ®┤ņŚŁ ļ░śņØæņŚÉ ļīĆĒĢ┤ ņé┤ĒÄ┤ļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
ņäĖĻĘĀ ņĖĪļ®┤
H. pyloriļŖö ņ▓śņØī Campylobacter ņåŹ ņäĖĻĘĀņ£╝ļĪ£ ļČäļźśļÉśņŚłņ£╝ļéś ļ»ĖņäĖĻĄ¼ņĪ░, 16S rRNA ņä£ņŚ┤, ņäĖĒżļ¦ē ņ¦Ćļ░®ņé░ ĻĄ¼ņä▒, ĒÜ©ņåī ņāØņä▒ļĀź, ļ░░ņ¢æ ĒŖ╣ņä▒, methylated menaquinone-6ņØś Ļ▓░ĒĢŹ ļō▒ņØä ĻĖ░ņżĆņ£╝ļĪ£ 1989ļģäņŚÉ ņāłļĪ£ņÜ┤ ĻĘĀņåŹņØĖ HelicobaterļĪ£ ņ×æļ¬ģļÉśņ¢┤ ņ×¼ļČäļźśļÉśņŚłļŗż. H. pyloriņØś ņ£ĀņĀäņ▓┤(genome) Ēü¼ĻĖ░ļŖö 1,643-1,736 kbņØ┤ļ®░ Campylobacter ĻĘĀņåŹņØś ņäĖĻĘĀĻ│╝ ņ£Āņé¼ĒĢ£ Ēü¼ĻĖ░ņØ┤Ļ│Ā ļīĆņןĻĘĀ ņ£ĀņĀäņ▓┤ņØś ņĢĮ 1/3ņĀĢļÅäņØś Ēü¼ĻĖ░ņØ┤ļŗż. H. pyloriļ¦īņØ┤ Ļ░Ćņ¦ĆļŖö ņ£ĀņĀäņ▓┤ņØś ĒŖ╣ņ¦Ģņ£╝ļĪ£ļŖö ņ▓½ņ¦Ė, ņÜ┤ļÅÖņä▒Ļ│╝ Ļ┤ĆļĀ©ļÉ£ ņ▓┤Ļ│äĻ░Ć ļ░£ļŗ¼ļÉśņ¢┤ ņ׳ņ¢┤ ĒÄĖļ¬©ņØś ĻĄ¼ņĪ░ļź╝ ļ¦īļōżĻ│Ā ņĪ░ļ”ĮĒĢśĻ│Ā, ņäĖĻĘĀņ▓┤ņÖĖļĪ£ ļČäļ╣äĒĢśļŖö Ļ▓āņØä ņĪ░ņĀłĒĢśļŖö ļŗ©ļ░▒ņ¦łņØ┤ 40Ļ░Ćņ¦Ć ņØ┤ņāüņØ┤ļŗż. ļæśņ¦Ė, ņ▓ĀļČä ĒØĪņłśļź╝ ņ£äĒĢ£ ņ▓┤Ļ│äĻ░Ć ņל ļ░£ļŗ¼ļÉśņ¢┤ ņ׳ļŗż. ņģŗņ¦Ė, ņĀ£ĒĢ£ ĒÜ©ņåīņØś endonuclease, methyltransferase, specificity subunit ņ£ĀņĀäņ×É ņ£Āņé¼ņä▒ņØä ņé┤ĒÄ┤ ļ│╝ ļĢī ņĀ£ĒĢ£Ļ│╝ ņłśļ│Ąņ▓┤Ļ│äĻ░Ć ņל ļ░£ļŗ¼ļÉśņ¢┤ ņ׳ļŗż. ļäĘņ¦Ė, ļČĆņ░®ņåīļĪ£ ņČöņĀĢļÉśļŖö ņ¦Ćņ¦łļŗ©ļ░▒Ļ│╝ ņÖĖļ¦ēļŗ©ļ░▒ņØ┤ ļ¦Äļŗż. H. pyloriĻ░Ć ņØ╝ļ░śņäĖĻĘĀņØ┤ ņĀĢņ░®ĒĢĀ ņłś ņŚåļŖö ņ£äņĀÉļ¦ēņŚÉņä£ ņāØņĪ┤ĒĢĀ ņłś ņ׳ļŖö Ļ▓āņØĆ ņØ┤ ņäĖĻĘĀņØ┤ ņ¦Ćļŗī ļÅģĒŖ╣ĒĢ£ ņĀüņØæĻĖ░ņĀä ļĢīļ¼ĖņØ┤ļŗż. H. pyloriĻ░Ć ņØĖņ▓┤ ņ£äņŗŁņØ┤ņ¦Ćņן ņĀÉļ¦ēņØś ņĀÉņĢĪņĖĄņŚÉ ņ╣©Ēł¼ĒĢśņŚ¼ ņ£äņĀÉļ¦ēņØś ļ░®ņ¢┤ĻĖ░ņĀäņØä ĻĄÉļ×ĆĒĢśĻ│Ā ĒÖ£ņä▒ņé░ņåīņŚÉ ļīĆĒĢ£ ļ░®ņ¢┤ ņ▓┤Ļ│äļź╝ ļ¼┤ļäłļ£©ļ”¼Ļ│Ā ņ¦łļ│æņØä ņØ╝ņ£╝ĒéżļŖö ņäĖĻĘĀĒĢÖņĀü ņØĖņ×ÉļōżņŚÉ ļīĆĒĢ┤ ņĢīņĢäļ│Ėļŗż.
ĒÄĖļ¬©
H. pyloriļŖö ņ£ä ņĀÉļ¦ēņŚÉņä£ ĒÖ£ļ░£ĒĢ£ ņÜ┤ļÅÖņä▒ņØä Ļ░Ćņ¦ĆĻ│Ā ņ׳ļŖöļŹ░, ņØ┤ļ¤¼ĒĢ£ Ļ░ĢļĀźĒĢ£ ņÜ┤ļÅÖņä▒ ļĢīļ¼ĖņŚÉ H. pyloriļŖö ņ£ä ņĀÉņĢĪņĖĄņØä ņ╣©Ēł¼ĒĢĀ ņłś ņ׳Ļ│Ā ņĀÉņĢĪņĖĄ ļé┤ņŚÉņä£ļÅä ņ×Éņ£ĀļĪŁĻ▓ī ņØ┤ļÅÖĒĢśļ®┤ņä£ ņ£ä ņĀÉļ¦ē ņāüĒö╝ņäĖĒżņØś ņĀæĒĢ®ļČĆņŚÉņä£ ņé┤ņĢäĻ░ł ņłś ņ׳ļŗż.
ļČĆņ░®ņåī(adhesin)
ļČĆņ░®ņåīļŖö ņØĖņ▓┤ ņĀÉļ¦ē Ēæ£ļ®┤ņŚÉņä£ Ļ░ÉņŚ╝ņØä ņØ╝ņ£╝ĒéżļŖö ļŗżņ¢æĒĢ£ ņäĖĻĘĀļōżņŚÉņä£ Ļ┤Ćņ░░ļÉśļŖö ņä▒ļČäņØ┤ļŗż. H. pyloriļŖö ņ£ĀņĀäņ▓┤ ņłśņżĆņŚÉņä£ļÅä ļŗżņ¢æĒĢ£ ĒĢŁņøÉ ļ│ĆņØ┤ļź╝ ņØ╝ņ£╝Ēé¼ ņłś ņ׳ļŖö ļ¼╝ņ¦łņØä ņāØņé░ĒĢśļŖö ņäĖĻĘĀņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ĒŖ╣Ē׳ sialic acid-specifc hemagglutinin(hpaA)Ļ│╝ lipid-binding ļō▒ņØĆ H. pyloriĻ░Ć ņ£ä ņĀÉļ¦ē ņāüĒö╝ņäĖĒżņŚÉ ņĀĢņ░®ĒĢśļŖö Ļ▓āņØä ļ¦żĻ░£ĒĢĀ Ļ▓āņ£╝ļĪ£ ņČöņĖĪĒĢśĻ│Ā ņ׳ļŗż.
ņĪ░ņ¦üļČäĒĢ┤ĒÜ©ņåī
H. pyloriĻ░Ć ļČäļ╣äĒĢśļŖö leucine aminopeptidaseĻ│╝ glycosulfaeaseļŖö Ļ░üĻ░ü ņĢīļČĆļ»╝Ļ│╝ mucinņØä ļČäĒĢ┤ĒĢ£ļŗż. ļśÉ H. pyloriļŖö phospholipase A1, A2, Cļź╝ ĒĢ®ņä▒ĒĢśļ®░, ĒĢ®ņä▒ļÉ£ phospholipase A1Ļ│╝ A2ļŖö ņäĖĻĘĀņ▓┤ņÖĖļĪ£ ļČäļ╣äļÉśĻ│Ā, phospholipase CļŖö ņäĖĻĘĀņ▓┤ļé┤ņŚÉ ņĪ┤ņ×¼ĒĢ£ļŗż. ņØ┤ phospholipaseļŖö ņ£äņĀÉļ¦ē ņāüĒö╝ņäĖĒżņØś ņäĖĒżļ¦ē lipid hydrophobic barrierļź╝ ĒīīĻ┤┤ĒĢśļŖö ņŚŁĒĢĀņØä ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉ£ļŗż.
Urease
H. pyloriļŖö ņÜöņåī(urea)ņÖĆņØś ņ╣£ĒÖöļĀźņØ┤ ļåÆĻ│Ā, ņÜöņåīļź╝ ņĢöļ¬©ļŗłņĢäņÖĆ COļĪ£ ļČäĒĢ┤ĒĢśļŖö ĒÜ©ņåīņØĖ urease ņāØņé░ĒĢ£ļŗż. H. pyloriĻ░Ć ņĀäņĀĢļČĆņŚÉ Ļ░ÉņŚ╝ņØä ņØ╝ņ£╝Ēéżļ®┤ ņØ┤ ļČĆņ£äņØś ņĀÉņĢĪņĖĄĻ│╝ ņāüĒö╝ņäĖĒż Ļ░ä ņĀæĒĢ®ļČĆņÖĆ Ēæ£ļ®┤ ņĀÉņĢĪņäĖĒżņŚÉ ļČĆņ░®ĒĢśņŚ¼ ņä£ņŗØĒĢ£ļŗż. ņ£äņןņØś ĻĄ¼ņĪ░ņÖĆ ĻĖ░ļŖźņØä Ļ│ĀļĀżĒĢĀ ļĢī H. pyloriļŖö ņ£äļé┤ņÜ®ļ¼╝ņØś pHļź╝ Ļ░Éņ¦ĆĒĢśĻ│Ā ņ£äņé░ ļČäļ╣äļź╝ ņĪ░ņĀłĒĢśļŖö ņĀäņĀĢļČĆņŚÉ ņä£ņŗØĒĢśļ»ĆļĪ£ H. pyloriņØś ureaseĻ░Ć ņÜöņåīļź╝ ļČäĒĢ┤ĒĢśņŚ¼ ņāØņä▒ļÉśļŖö ņĢöļ¬©ļŗłņĢäĻ░Ć ņĀäņĀĢļČĆņØś pHļź╝ ņāüņŖ╣ņŗ£Ēéżļ®┤ ņĀäņĀĢļČĆņØś pH Ļ░Éņ¦ĆļŖźņØ┤ Ļ░ÉņåīļÉ£ļŗż. H. pyloriĻ░Ć ņØĖņ¦Ćņ¦łņØä ļČäĒĢ┤ĒĢśĻ│Ā, ureaseņŚÉ ņØśĒĢ£ ņĢöļ¬©ļŗłņĢäņÖĆ ĻĖ░ĒāĆ ņäĖĻĘĀņé░ļ¼╝ļōżņØ┤ ņāüĒö╝ņäĖĒżļź╝ ņåÉņāüņŗ£Ēé©ļŗż. ļśÉĒĢ£ ņÜ┤ļÅÖņä▒ņØ┤ Ļ░ĢĒĢ£ H. pyloriĻ░Ć ņĀÉņĢĪņĖĄņØä ņØ┤ļÅÖĒĢśļ®┤ņä£ ļČäļ╣äĒĢśļŖö mucus-degrading glycosulfataseĻ░Ć ņĀÉņĢĪņØä ļČäĒĢ┤ĒĢśļ®┤ ņĀÉņĢĪņĖĄņØś Ļ▓®ņ×É ņ▓┤Ļ│äĻ░Ć ĻĄÉļ×ĆļÉśņ¢┤ ņ£ä ņĀÉļ¦ē ļ░®ņ¢┤ ņĀäņ▓┤Ļ░Ć ļČĢĻ┤┤ļÉ£ļŗż. ņØ┤ņ¢┤ņä£ acid-pepsin Ļ│ĄĻ▓®ņØĖņ×ÉĻ░Ć Ļ│Āņ£ĀĒīÉ(lamina propria)ņØä Ļ│ĄĻ▓®ĒĢśļŖö ļ╣äĻ░ĆņŚŁņĀüņØĖ ņĢģņł£ĒÖśņØ┤ ļ░£ņāØĒĢ£ļŗż. ĻĘĖļĀćĻ▓ī ļÉśļ®┤ ņ£ä ņĀÉļ¦ēņØś Ļ│Āņ£ĀĒīÉņŚÉ ļČäĒżĒĢśļŖö ļ╣äļ¦īņäĖĒżĻ░Ć Ē׳ņŖżĒāĆļ»╝ņØä ļČäļ╣äĒĢśĻ│Ā, ļ▓ĮņäĖĒżĻ░Ć Ļ│ĄĻ▓® ņØĖņ×ÉņØĖ HCIņØä Ļ│╝ļČäļ╣äĒĢśļÅäļĪØ ņ£ĀļÅäĒĢśņŚ¼ ņŗŁņØ┤ņ¦Ćņן ņĀÉļ¦ēņŚÉ gastric metaplasiaļź╝ ņ£Āļ░£ņŗ£Ēé©ļŗż.
ļÅģņä▒ņØĖņ×É
cag Pathogenicity Island (cag PAI)ņÖĆ CagAļŗ©ļ░▒: H. pyloriņØś ņ£ĀņĀäņ▓┤ņŚÉļŖö ļ¦ÄņØĆ ņ£ĀņĀäņĀü ļŗżĒśĢņä▒ņØ┤ ņĪ┤ņ×¼ĒĢśļŖöļŹ░, ĻĘĖ ņżæ cagPAIĻ░Ć ņל ņĢīļĀżņĀĖ ņ׳ļŗż[27]. cag PAIļŖö 40-kbņØś DNAļĪ£ ņĄ£ņóģ ņŻ╝ņÜö ņé░ļ¼╝ņØĆ CagAļŗ©ļ░▒ņØ┤ļŗż. ņä£ĻĄ¼ņŚÉņä£ļŖö H. pylori ĻĘĀņŻ╝ņØś ņĢĮ 60%ņŚÉņä£ cag PAIĻ░Ć ņ¢æņä▒ņØĖ ļ░śļ®┤ ņÜ░ļ”¼ļéśļØ╝ļź╝ ĒżĒĢ©ĒĢ£ ļÅÖņĢäņŗ£ņĢäņŚÉņä£ļŖö 100% ņ¢æņä▒ņØ┤ļŗż. cag PAI ņ¢æņä▒ H. pyloriņŚÉ Ļ░ÉņŚ╝ņØ┤ ļÉśļ®┤, cag PAI ņØīņä▒ H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉ£ Ļ▓ĮņÜ░ņŚÉ ļ╣äĒĢ┤ ņ£äņČĢņä▒ ņ£äņŚ╝ņØ┤ ļŹö ņŗ¼ĒĢśĻ▓ī ņØ╝ņ¢┤ļéśĻ│Ā ņ£äņäĀņĢö ļ░£ņāØņØś ņ£äĒŚśļÅäĻ░Ć ļŹö ļåÆļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż[28].
CagAļŗ©ļ░▒ņØĆ 120-140 kDa Ēü¼ĻĖ░ņØś ļŗ©ļ░▒ņ£╝ļĪ£, H. pyloriĻ░Ć ņ£ä ņĀÉļ¦ēņØś ņāüĒö╝ņäĖĒżņŚÉ ļČĆņ░®ĒĢśļ®┤ type IV secretion systemņØä ĒåĄĒĢ┤ ņłÖņŻ╝ņØś ņāüĒö╝ņäĖĒż ņĢłņ£╝ļĪ£ ņØ┤ļÅÖĒĢśņŚ¼ glutamine-proline-isoleucine-tyrosine-alanine motifņŚÉņä£ ņØĖņé░ĒÖöļÉśņ¢┤ ŌĆ£hummingbird phenomenonŌĆØņ£╝ļĪ£ ņĢīļĀżņ¦ä ņäĖĒż ĒśĢĒā£ņØś ļ│ĆĒÖöļź╝ ņ£Āļ░£ņŗ£Ēé©ļŗż[29,30]. ņØĖņé░ĒÖöĒĢśņ¦Ć ņĢŖņØĆ CagA ļŗ©ļ░▒ļÅä ņĀäņ£ä(translocation)ņØä ĒåĄĒĢśņŚ¼ ņ¦łļ│æ ļ░£ņāØņŚÉ Ļ┤ĆņŚ¼ĒĢ£ļŗż[31]. ņ”ē, ╬▓-cateninņØś ņØ┤ņāü ĒÖ£ņä▒ĒÖö, ņäĖĒżņÖĆ ņäĖĒżļź╝ ņŚ░Ļ▓░ĒĢśļŖö ĻĄ¼ņĪ░ļ¼╝ņØś ĒīīĻ┤┤, ņäĖĒżņØś ĻĘ╣ņä▒ ņāüņŗż ļō▒ņØä ņ£Āļ░£ĒĢ£ļŗż. CagAļŗ©ļ░▒ņØĆ ņåīĒÖöņä▒ĻČżņ¢æņØś ļ░£ņāØĻ│╝ļÅä ļ░ĆņĀæĒĢ£ Ļ┤ĆĻ│äĻ░Ć ņ׳ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż[32].
VacA ļÅģņåī: VacAļŗ©ļ░▒ņØĆ 87 kDa Ēü¼ĻĖ░ņØś ļŗ©ļ░▒ņ£╝ļĪ£ H. pyloriņØś vacA ņ£ĀņĀäņ×É ļ░£Ēśä ņé░ļ¼╝ņØ┤ļŗż[27]. VacAļŗ©ļ░▒ņØĆ ņłÖņŻ╝ņäĖĒżņØś Ļ│ĄĒżĒÖö(vacuolation)ļź╝ ņ£Āļ░£ĒĢśļŖö ņäĖĒżļÅģņåī(cytotoxin)ļĪ£, H. pyloriņŚÉ ļīĆĒĢ£ ņłÖņŻ╝ņØś TņäĖĒż ļ░śņØæņØä ņ¢ĄņĀ£ĒĢ©ņ£╝ļĪ£ņŹ© Ļ░ÉņŚ╝ņØ┤ ņ¦ĆņåŹļÉśļÅäļĪØ ĒĢ£ļŗż[33]. vacA ņ£ĀņĀäņ×ÉļŖö H. pylori ĻĘĀņŻ╝ ļīĆļČĆļČäņŚÉ ņĪ┤ņ×¼ĒĢśļ®░ ņŗĀĒśĖļČĆņ£ä(s) ļśÉļŖö ņżæĻ░äļČĆņ£ä(m) ļō▒ ņĪ┤ņ×¼ĒĢśļŖö ņ£äņ╣śņŚÉ ļö░ļØ╝ s1, s2, m1, m2 ļō▒ņØś ņĢäĒśĢņØ┤ ņ׳ņ£╝ļ®░, ņĢäĒśĢņŚÉ ļö░ļØ╝ ņäĖĒżļÅģņä▒ņØ┤ ļŗżņ¢æĒĢśĻ▓ī ļéśĒāĆļé£ļŗż[34]. ņØ┤ ņżæ vacA s1/m1 ņĢäĒśĢņØ┤ Ļ░Ćņן ņäĖĒżļÅģņä▒ņØ┤ Ļ░ĢĒĢśĻ│Ā, ņÜ░ļ”¼ļéśļØ╝ļź╝ ĒżĒĢ©ĒĢ£ ļīĆļČĆļČäņØś ļÅÖņĢäņŗ£ņĢäņØĖņŚÉņä£ ļ░£Ļ▓¼ļÉ£ļŗż[35]. VacA ļŗ©ļ░▒ņØĆ ņ£ä ņāüĒö╝ņäĖĒżņØś ĒśĢĒā£ļź╝ ļ│ĆĒśĢņŗ£ņ╝£ ļ░®ļ▓ĮĒÜ©Ļ│╝ļź╝ ĒīīĻ┤┤ĒĢśĻ│Ā ņŚ╝ņ”Øļ░śņØæņØä ņĪ░ņĀłĒĢ£ļŗż. ļśÉĒĢ£ ņäĖĒż ļé┤ ņŚöļÅäņå£(endosome) ĻĄ¼ĒÜŹ, ļ»ĖĒåĀņĮśļō£ļ”¼ņĢäņØś ņÖĖļ¦ē ļō▒ņØä ĒīīĻ┤┤ĒĢśņŚ¼ ņäĖĒżņØś Ļ│ĄĒżĒÖöļź╝ ņØ╝ņ£╝ĒéżĻ│Ā, caspase-8, caspase-9ļź╝ ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ ņäĖĒżņé¼ļ®ĖņØä ņ£ĀļÅäĒĢ£ļŗż[36]. VacAļŗ©ļ░▒ņØ┤ Ļ▓░ĒĢ®ĒĢśļŖö ņ£ä ņāüĒö╝ņäĖĒżņØś ņłśņÜ®ņ▓┤ ņżæ receptor type protein tyrosine phosphataseļŖö ņäĖĒżņØś ņ”ØņŗØ, ļČäĒÖö, ļČĆņ░® ļō▒Ļ│╝ Ļ┤ĆļĀ©ņØ┤ ņ׳ņ£╝ļ®░ ņØ┤ļź╝ ĒåĄĒĢ┤ ņåīĒÖöņä▒ĻČżņ¢æņØä ņØ╝ņ£╝ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[37].
ņÖĖļ¦ēļŗ©ļ░▒(outer membrane protein): H. pyloriĻ░Ć ņ£ä ņāüĒö╝ņäĖĒżņŚÉ ņל ļČĆņ░®ĒĢśĻĖ░ ņ£äĒĢ┤ņä£ļŖö ļČĆņ░®ņåīļ┐Éļ¦ī ņĢäļŗłļØ╝ ņÖĖļ¦ēļŗ©ļ░▒ļÅä ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢ£ļŗż. H. pyloriņØś ņÖĖļ¦ēļŗ©ļ░▒ņŚÉļŖö blood group antigen binding adhesin, sialic acid-binding adhesin, outer inflammatory protein, duodenal ulcer promoting gene, flagella ļō▒ņØ┤ ĒżĒĢ©ļÉśņ¢┤ ņ׳Ļ│Ā, ņØ┤ļ¤¼ĒĢ£ ļŗ©ļ░▒ņØĆ ļŗżņ¢æĒĢ£ ņ£äņןĻ┤Ć ņ¦łĒÖśĻ│╝ ņŚ░Ļ┤ĆņØ┤ ņ׳ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż.
ļ®┤ņŚŁ ņĖĪļ®┤
H. pyloriĻ░Ć ņāüĒö╝ņäĖĒżņĖĄņŚÉ ņĀĢņ░®ĒĢśļ®┤ ņ£äņāüĒö╝ņäĖĒżļĪ£ļČĆĒä░ ņŗĀĒśĖĻ░Ć ņāØņä▒ļÉśĻ│Ā, ĻĘĖ Ļ▓░Ļ│╝ ņØ╝ļĀ©ņØś ļ®┤ņŚŁ ļ░śņØæņØ┤ ņ£Āļ░£ļÉ£ļŗż(Fig. 3). ļö░ļØ╝ņä£ H. pylori Ļ░ÉņŚ╝ņŚÉ ņØśĒĢ£ ņ┤łĻĖ░ņØś ļ®┤ņŚŁļ░śņØæņØĆ ņŻ╝ļĪ£ ņ£äņāüĒö╝ņäĖĒżĻ░Ć ļŗ┤ļŗ╣ĒĢśĻ│Ā ņØ┤ĒøäņŚÉļŖö ņĀÉļ¦ē ņāüĒö╝ņäĖĒżņĖĄ ĒĢśļČĆļĪ£ ļ¬©ņŚ¼ļōĀ ļ®┤ņŚŁņäĖĒżņŚÉ ņØśĒĢ┤ ņĪ░ņĀłļÉ£ļŗż[38]. ņØ┤ļ¤¼ĒĢ£ ļ®┤ņŚŁņäĖĒżņŚÉ ņØśĒĢ┤ ļéśĒāĆļéśļŖö ļ░śņØæņØĆ ņäĖĒżņä▒ ļ®┤ņŚŁļ░śņØæ(cellular immune response)Ļ│╝ ņ▓┤ņĢĪņä▒ ļ®┤ņŚŁļ░śņØæ(humoral immune response)ņ£╝ļĪ£ ĻĄ¼ņä▒ļÉ£ļŗż.
ņäĀņ▓£ ļ®┤ņŚŁļ░śņØæ(innate immune response)
H. pyloriļŖö ņ£äļé┤ļĪ£ ļōżņ¢┤ņś© Ēøä ņĀÉņĢĪņĖĄņØä ņ╣©Ēł¼ĒĢśĻ│Ā ņ£äļé┤Ļ░ĢņŚÉ ļ╣äĒĢ┤ņä£ ņé░ļÅäĻ░Ć ļåÆņ¦Ć ņĢŖņØĆ ņ£äņāüĒö╝(pH 5-6)Ļ╣īņ¦Ć ļÅäļŗ¼ĒĢ£ļŗż. ņ£ä ņāüĒö╝ņäĖĒż Ēæ£ļ®┤ņŚÉ ļÅäļŗ¼ĒĢ£ ņäĖĻĘĀņŚÉ ļīĆĒĢ┤ ņäĀņ▓£ņĀü ļ®┤ņŚŁļ░śņØæņØ┤ ņØ╝ņ¢┤ļé£ļŗż. ņØ┤ļĢī bacterial pathogen-associated molecular patterns(lipopolysaccharide [LPS], flagellin, peptidoglycan)ņÖĆ pattern recognition receptorņØĖ nucleotide-binding oligomerization domain protein I (Nod1)Ļ│╝ Toll-like receptors (TLRs)ņÖĆ Ļ░ÖņØĆ ņäĀņ▓£ņĀü ļ®┤ņŚŁ ņ▓┤Ļ│äņØś ļŗżņ¢æĒĢ£ ņÜöņåīļōżņØ┤ ļ®┤ņŚŁ ļ░śņØæņØä ņ£ĀļÅäĒĢśĻ│Ā, ĒĢŁņäĖĻĘĀņä▒ ļŗ©ļ░▒ņ¦łņØ┤ ļČäļ╣äļÉ£ļŗż[39].
TLRsļŖö ņ£ä ņāüĒö╝ Ēæ£ļ®┤ņŚÉņä£ ļ░£Ļ▓¼ļÉśļ®░ ņøÉĒśĢņ¦łļ¦ē(plasma membrane) Ēś╣ņØĆ ļ”¼ļ│┤ņóĆ ņåīĒż(ribosomal vesicles)ļéś ņŚöļÅäņóĆ ņåīĒż(endosomal vesicles)ņÖĆ ņŚ░Ļ┤ĆņØ┤ ņ׳ļŖö ļ░śļ®┤ņŚÉ, Nod1ņØĆ ņäĖĒżņ¦ł ņĢłņŚÉ ņĪ┤ņ×¼ĒĢ£ļŗż[40]. cagPAIņŚÉ ņØśĒĢ┤ ņ£ä ņāüĒö╝ņŚÉ ņĀäļŗ¼ļÉ£ peptidoglycan fragmentsļŖö ņäĖĒżņ¦ł ņäĀņ▓£ņĀü ļ®┤ņŚŁļČäņ×ÉņØĖ Nod1ņŚÉ ņØśĒĢ┤ ņØĖņŗØļÉśļ®░, ņØ┤ļŖö ņāüĒö╝ņäĖĒżņŚÉņä£ H. pyloriļź╝ ņ¦üņĀæ ņĀ£Ļ▒░ĒĢśļŖö ņŚŁĒĢĀņØä ĒĢ£ļŗż[41]. ļśÉĒĢ£ ņ£ä ņāüĒö╝ņäĖĒżļŖö ╬▒-defensin ļ░Å ╬▓-defensinĻ│╝ cathelicidin LL-37Ļ│╝ Ļ░ÖņØĆ ļŗżņ¢æĒĢ£ ĒĢŁņäĖĻĘĀņä▒ ļŗ©ļ░▒ņ¦łļōżņØä ņāØņé░ĒĢ£ļŗż[42,43]. ņØ┤ļ¤¼ĒĢ£ ļŗ©ļ░▒ņ¦łļōżņØĆ ņäĖĻĘĀņØś ņä▒ņןņØä ņ¢ĄņĀ£ĒĢśņŚ¼ ņäĖĻĘĀņŚÉ ļīĆĒĢ£ ļ░®ņ¢┤ņ×æņÜ®ņØä ĒĢ£ļŗż. H. pyloriļŖö TLRsļź╝ ĒåĄĒĢ┤ņä£ ņłÖņŻ╝ ņäĖĒżņŚÉņä£ ņŚ╝ņ”Ø ņ£Āļ░£ ņ£ĀņĀäņ×ÉņØś ļ░£ĒśäņØä ņ£ĀļÅäĒĢ£ļŗż. TLRsļŖö ļ¦ēĻ┤ĆĒåĄļŗ©ļ░▒ņ¦ł(transmembrane protein)ņØś ņ¦æĒĢ®ņ▓┤ļĪ£ TLR4ļŖö ņäĖĻĘĀņØś LPSļź╝, TLR2ļŖö peptidoglycanņØä, TLR5ļŖö flagellinņØä ņØĖņ¦ĆĒĢ£ļŗż. ĒÖ£ņä▒ĒÖöļÉ£ TLRsļŖö nuclear factor kappa Bļź╝ ĒÖ£ņä▒ĒÖöĒĢśņŚ¼ ņŚ╝ņ”Øņä▒ cytokine ņ£ĀņĀäņ×Éļź╝ ņĀäņé¼ĒĢ£ļŗż[39].
ChemokinesņÖĆ ņŚ╝ņ”ØņäĖĒż
H. pyloriņŚÉ ļīĆĒĢ£ ņäĀņ▓£ņĀü ļ®┤ņŚŁļ░śņØæņŚÉ ņØśĒĢ┤ ļČäļ╣äļÉśļŖö cytokinesļŖö ņ£äņŚ╝ ļ░£ņāØņØä ņ┤ēņ¦äņŗ£ĒéżĻ│Ā ņżæņä▒ĻĄ¼ņÖĆ ļŗżļźĖ ļ®┤ņŚŁ ņäĖĒżļōżņØś ņ£Āņ×ģņØä ņ£ĀļÅäĒĢ£ļŗż[39]. ņżæņä▒ĻĄ¼ņØś ņ╣©ņ£żņØĆ H. pyloriĻ░Ć ņ£äņŚÉ ņ▓śņØī ņä£ņŗØĒĢĀ ļĢī Ļ┤Ćņ░░ļÉśļ®░, ņä▒ņØĖĻĖ░ņŚÉ ņ¦ĆņåŹļÉśļŖö Ļ░ÉņŚ╝ņŚÉņä£ļÅä Ļ┤Ćņ░░ĒĢĀ ņłś ņ׳ļŗż. H. pyloriņŚÉ ņØśĒĢ┤ ņ£ĀļÅäļÉ£ interleukin (IL-8)Ļ│╝ growth related oncogene ╬▒ņÖĆ Ļ░ÖņØĆ cytokinesļŖö ņżæņä▒ĻĄ¼ņØś ĒÖ£ņä▒ĒÖö ļ░Å ņØ┤ļÅÖņØä ņĪ░ņĀłĒĢ£ļŗż[44]. H. pyloriņØś VacAņÖĆ neutrophil activating protein (Hp-NAP)ņØĆ ļ╣äļ¦īņäĖĒżļź╝ ĒÖ£ņä▒ĒÖöņŗ£ņ╝£ ņŚ╝ņ”ØņØä ļŹöņÜ▒ ņ”ØĻ░Ćņŗ£Ēé©ļŗż. Hp-NAPļŖö TLR2ņŚÉ ņ×æņÜ®ĒĢśņŚ¼ ņżæņä▒ĻĄ¼ņÖĆ ļŗ©ĒĢĄĻĄ¼ņŚÉņä£ IL-12ņÖĆ IL-23ņØś ļ░£ĒśäņØä ņ£ĀļÅäĒĢśņŚ¼ ņ£ä ņāüĒö╝ņŚÉņä£ H. pylori-ĒŖ╣ņØ┤ņĀü Th1 ļ®┤ņŚŁļ░śņØæņØä ņ┤ēņ¦äņŗ£Ēé©ļŗż. IL-12ļŖö na├»ve Th ņäĖĒżĻ░Ć Th1 phenotypeņ£╝ļĪ£ ļČäĒÖöĒĢśļŖö ļŹ░ Ļ┤ĆņŚ¼ĒĢśļ®░, Ļ░ĢļĀźĒĢ£ ņŚ╝ņ”Ø ņ£Āļ░£ cytokineņ£╝ļĪ£ ņ×æņÜ®ĒĢ£ļŗż. Hp-NAPņÖĆ Ļ░ÖņØĆ ņäĖĻĘĀņØś ļÅģņä▒ ņØĖņ×ÉļōżņØĆ ņżæņä▒ĻĄ¼ļź╝ ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻĖ░ļÅä ĒĢśņ¦Ćļ¦ī, ņśżĒ׳ļĀż ņżæņä▒ĻĄ¼ņØś ĻĖ░ļŖźņØä ņĀĆĒĢ┤ĒĢśĻĖ░ļÅä ĒĢ£ļŗż. ņŗØĒż(phagosome) ĒśĢņä▒ņØä ņ¦ĆņŚ░ĒĢśņŚ¼ ļīĆņŗØņäĖĒżņŚÉ ņØśĒĢ£ ņĀ£Ļ▒░ļź╝ ĒÜīĒö╝ĒĢśĻ│Ā, ņāüĒö╝ ņäĖĒżņÖĆ ņłśņ¦Ćņāü ņäĖĒż(dentritic cell) ļé┤ņŚÉņä£ ņ”ØņŗØĒĢĀ ņłś ņ׳ļŗż. H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉ£ ņ£ä ņāüĒö╝ņŚÉņä£ļŖö interferon ╬│, tumor necrosis factor-╬▒ (TNF-╬▒)ņÖĆ IL (IL-1╬▓, IL-6, IL-7, IL-8, IL-10, IL-18) ļō▒ ļ¦ÄņØĆ cytokinesņØś ņ¢æņØ┤ ņ”ØĻ░ĆļÉśņ¢┤ ņ׳ļŗż. ņØ┤ļ¤¼ĒĢ£ cytokinesņØś ļīĆļČĆļČäņØĆ ņŚ╝ņ”ØņØä ņ£Āļ░£ĒĢśņ¦Ćļ¦ī, IL-10ņØĆ ņŚ╝ņ”Øņä▒ Th1 ļ®┤ņŚŁļ░śņØæņØä ņ¢ĄņĀ£ĒĢśļŖö ļ®┤ņŚŁ ņĪ░ņĀłļĀźņØä Ļ░Ćņ¦äļŗż.
ņĀüņØæ ļ®┤ņŚŁļ░śņØæ(adaptive immune response)
H. pyloriļŖö Ļ░ĢļĀźĒĢ£ ņ▓┤ņĢĪņä▒(humoral immunity) ļ░Å ņäĖĒżļ¦żĻ░£ņä▒ ļ®┤ņŚŁ(cell-mediated immunity)ņØä ņ£ĀļÅäĒĢ£ļŗż. ņØ┤ļŖö Ļ░ÉņŚ╝ Ēøä ļ¼┤ņ”Øņāü Ļ▓ĮĻ│╝ņŚÉļŖö ļÅäņøĆņØ┤ ļÉśņ¦Ćļ¦ī ņĪ░ņ¦üņØś ņåÉņāüņØä ņ£Āļ░£ĒĢĀ ņłś ņ׳ļŗż. ņäĖĻĘĀņØĆ ņłśņ¦Ćņāü ņäĖĒż, TņäĖĒż ļ░Å BņäĖĒżņÖĆ ņāüĒśĖņ×æņÜ®ĒĢśņŚ¼ ĒÜŹļōØņä▒ ļ®┤ņŚŁļ░śņØæ(acquired immune response)ņØä ĒśĢņä▒ĒĢ£ļŗż. ņłśņ¦Ćņāü ņäĖĒżļŖö ņäĖĻĘĀņØä ĒżņŗØĒĢśĻ│Ā ņ▓śļ”¼ĒĢśņŚ¼ ņŻ╝ņĪ░ņ¦üņĀüĒĢ®ļ│ĄĒĢ®ņ▓┤(major histocompatibility complex)ļź╝ Ļ░Ćņ¦ĆĻ▓ī ļÉśļŖöļŹ░, ņØ┤ļŖö ĒŖ╣ņØ┤ TņäĖĒżĻ░Ć ļ░śņØæĒĢĀ ņłś ņ׳ļÅäļĪØ peptideļź╝ ņĀäļŗ¼ĒĢ£ļŗż. ļśÉĒĢ£ H. pyloriļŖö ņłśņ¦Ćņāü ņäĖĒżļź╝ ņ×ÉĻĘ╣ĒĢśņŚ¼ IL-6, IL-8, IL-10 ļ░Å IL-12ņÖĆ Ļ░ÖņØĆ ļŗżņ¢æĒĢ£ cytokinesļź╝ ļ░£Ēśäņŗ£Ēé©ļŗż.
BņäĖĒżņŚÉņä£ CagAņØś ļ░£ĒśäņØĆ Janus kinase-signal transducer and activator of transcription ņŗĀĒśĖņĀäļŗ¼ņØä ņ¢ĄņĀ£ĒĢ©ņ£╝ļĪ£ņŹ© IL-3-dependent BņäĖĒż ņ”ØņŗØņØä Ļ░Éņåīņŗ£Ēé©ļŗż. ņØ┤ļĪ£ ņØĖĒĢ┤ ĒĢŁņ▓┤ ņāØņä▒ņØ┤ Ļ░ÉņåīĒĢśĻ│Ā cytokinesņØś ļ░£ĒśäņØ┤ ņ¢ĄņĀ£ļÉ£ļŗż. H. pylori Ļ░ÉņŚ╝ņŚÉ ļīĆĒĢ£ ļ®┤ņŚŁļ░śņØæņŚÉņä£ļŖö TņäĖĒżĻ░Ć ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢ£ļŗż. Th1/Th2 ļ®┤ņŚŁļ░śņØæņØś ĻĘĀĒśĢņØĆ H. pyloriņÖĆ ņŚ░Ļ┤ĆļÉ£ ņ¦łĒÖśņØś ļ│æņØĖĻ│╝ ņłÖņŻ╝ņØś ļ░®ņ¢┤ ĻĖ░ņĀäņŚÉņä£ ņżæņÜöĒĢśļŗż. Th1 ļ®┤ņŚŁļ░śņØæņØ┤ ņÜ░ņäĖĒĢśļ®┤ ņĪ░ņ¦ü ņåÉņāüņØä ņØ╝ņ£╝Ēéżņ¦Ćļ¦ī, Th2 ļ®┤ņŚŁļ░śņØæņØĆ ņ£ä ņŚ╝ņ”Ø ļ░śņØæņŚÉ ļīĆĒĢ£ ļ░®ņ¢┤ņ×æņÜ®ņØä ĒĢ£ļŗż. ņØ┤ņ▓śļ¤╝ TņäĖĒżņØś ņäĖĒż ļÅģņä▒ņØĆ H. pylori Ļ░ÉņŚ╝ņØś ņ×äņāü Ļ▓ĮĻ│╝ņŚÉ ņżæņÜöĒĢ£ ņśüĒ¢źņØä ņżĆļŗż. H. pyloriļŖö TņäĖĒż ņ”ØņŗØņØä ņ¢ĄņĀ£ņŗ£Ēé┤ņ£╝ļĪ£ņŹ© Ļ░ÉņŚ╝ņØä ņ¦ĆņåŹņŗ£Ēéżļ®░, TņäĖĒż ņØśņĪ┤ņä▒ BņäĖĒż ņ”ØņŗØņØä ņØ╝ņ£╝ņ╝£ ņØ╝ļČĆ ĒÖśņ×ÉņŚÉņä£ ņ£ä ļ│ĆņŚ░ļČĆ BņäĖĒż ļ”╝ĒöäņóģņØä ņØ╝ņ£╝ĒéżĻĖ░ļÅä ĒĢ£ļŗż. ļ®┤ņŚŁ ņĪ░ņĀł ņäĖĒż(ņŻ╝ļĪ£ TregņäĖĒż)ļŖö ĒÜŹļōØņä▒ ļ®┤ņŚŁļ░śņØæņØä ņĪ░ņĀłĒĢśļŖö ļŹ░ ņżæņÜöĒĢśļ®░, ņ£ä ņāüĒö╝ņŚÉņä£ ļ│æļ”¼ĒĢÖņĀüņØĖ ļ│ĆĒÖöļź╝ ņĪ░ņĀłĒĢĀ ļ┐Éļ¦ī ņĢäļŗłļØ╝, Ļ░ÉņŚ╝ņØś ļ¦īņä▒ĒÖöļź╝ ņØ╝ņ£╝ĒéżĻĖ░ļÅä ĒĢ£ļŗż. ņ▓┤ņĢĪņä▒ ļ®┤ņŚŁļ░śņØæ(humoral immune response) ļśÉĒĢ£ H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉ£ Ļ▒░ņØś ļ¬©ļōĀ Ļ░£ņ▓┤ņŚÉņä£ ļ░£ņāØĒĢ£ļŗż. ĒŖ╣ņØ┤ Ēśłņ▓Ł IgM ĒĢŁņ▓┤ļŖö ļ│┤ĒåĄ Ļ░ÉņŚ╝ Ēøä ņ▓½ ņłśĻ░£ņøö ļÅÖņĢł ļéśĒāĆļé£ļŗż. Ēśłņ▓Ł IgAņÖĆ IgG ĒĢŁņ▓┤ļŖö H. pyloriņØś ļŗżļźĖ ĒĢŁņøÉĻ▓░ņĀĢļČĆņ£ä(antigenic determinant)ņŚÉ ļ░śņØæĒĢśņŚ¼ ļéśĒāĆļéśĻ│Ā, ĒŖ╣ņØ┤ ļČäļ╣äņä▒ IgA ĒĢŁņ▓┤ļŖö ĻĄŁņåīņĀüņØĖ ļ®┤ņŚŁ ļ░śņØæņ£╝ļĪ£ņä£ ņ£äņĢĪņŚÉ ņĪ┤ņ×¼ĒĢ£ļŗż. ĒĢśņ¦Ćļ¦ī ņØ┤ļ¤¼ĒĢ£ ĒĢŁņ▓┤ļōżņØś ĒÜ©Ļ│╝ņŚÉ ļīĆĒĢ┤ņä£ļŖö ļģ╝ļ×ĆņØ┤ ņ׳ļŗż.
ļ®┤ņŚŁļ░śņØæĻ│╝ ņ×äņāüņ¢æņāü
H. pylori Ļ░ÉņŚ╝ņŚÉ ņØśĒĢ£ ņ£ä ņŚ╝ņ”ØņØĆ cagPAI, vacA, dupA ļō▒ ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņäĖĻĘĀņØś ļÅģņä▒ ņØĖņ×ÉļōżņŚÉ ņØśĒĢ┤ ņ”ØĻ░ĆļÉ£ļŗż. ĻĘĖļ¤¼ļéś, ņäĖĻĘĀņŚÉ ļīĆĒĢ£ ļ®┤ņŚŁļ░śņØæņØĆ ļ¦żņÜ░ ļ│Ąņ×ĪĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ ņłÖņŻ╝ņŚÉ ļö░ļØ╝ ņŚ╝ņ”ØņØś ņĀĢļÅäĻ░Ć ļŗ¼ļØ╝ņ¦ł ņłś ņ׳ļŗż. Cytokinesļź╝ codingĒĢśļŖö ņ£ĀņĀäņ×É ļŗżĒśĢņä▒ņØĆ Ļ░ÉņŚ╝ņŚÉ ņØśĒĢ£ ņ×äņāü ļ░£ĒśäņŚÉ ļ│ĆĒÖöļź╝ ņżä ņłś ņ׳ļŗż. ĒŖ╣Ē׳ IL-1╬▓ņÖĆ TNF-╬▒ņØś ņ”ØĻ░Ć, IL-10ņØś Ļ░ÉņåīņÖĆ ņŚ░Ļ┤ĆļÉ£ ņ£ĀņĀäņ×É ļŗżĒśĢņä▒ņØĆ ņ£äņČĢņä▒ ņ£äņŚ╝Ļ│╝ ņ£äņĢöņØś ņ£äĒŚśļÅäļź╝ ņ”ØĻ░Ćņŗ£Ēé©ļŗżĻ│Ā ņĢīļĀżņĀĖ ņ׳ļŗż.
Ļ▓░ ļĪĀ
ņÜ░ļ”¼ļéśļØ╝ H. pylori ĒĢŁņ▓┤ņŚÉ ļīĆĒĢ£ Ēśłņ▓ŁĒĢÖņĀü ņ£Āļ│æļźĀņØĆ ņĢĮ 50%ļĪ£ ĻĄŁļ»╝ņØś ļ░śņłśĻ░Ć Ļ░ÉņŚ╝ļÉśņ¢┤ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉśļ®░ ĒŖ╣Ē׳ 16ņäĖ ņØ┤ņāüņØś ņä▒ņØĖņŚÉņä£ļŖö ņĢĮ 2/3Ļ░Ć Ļ░ÉņŚ╝ļÉśņ¢┤ ņ׳ļŗż. ņÜ░ļ”¼ļéśļØ╝ņØś H. pylori Ļ░ÉņŚ╝ļźĀņØĆ Ļ▓ĮņĀ£ņä▒ņןĻ│╝ ņāØĒÖ£ĒÖśĻ▓ĮņØś Ļ░£ņäĀ ļō▒ņ£╝ļĪ£ Ē¢źĒøä Ļ░ÉņåīĒĢĀ Ļ▓āņ£╝ļĪ£ ņśłņāüļÉ£ļŗż. ĻĘĖļ¤¼ļéś ņĀäĻĄŁņĀü ĻĘ£ļ¬©ņØś ņŚŁĒĢÖņŚ░ĻĄ¼Ļ░Ć ņĀĢĻĖ░ņĀüņ£╝ļĪ£ ņŗ£Ē¢ēļÉśņ¢┤ ļ│┤ļŗż Ļ░ØĻ┤ĆņĀüņØĖ ņ×ÉļŻīļź╝ ļ░öĒāĢņ£╝ļĪ£ H. pyloriņŚÉ ļīĆĒĢ£ ņ¦äļŻī ņ¦Ćņ╣©ņØä ļ¦īļōż ĒĢäņÜöĻ░Ć ņ׳ļŗż. H. pyloriņŚÉ Ļ░ÉņŚ╝ļÉśļ®┤ ņäĖĻĘĀ ļÅģņä▒ņØĖņ×ÉņØś ņ£ĀņĀäņĀü ļŗżĒśĢņä▒Ļ│╝ ņØ┤ņŚÉ ļīĆĒĢ£ ņłÖņŻ╝ņØś ļ®┤ņŚŁļ░śņØæņØ┤ ņāüĒśĖ ņ×æņÜ®ņØä ĒĢśņŚ¼ ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņ£äņן ņ¦łĒÖśņØä ņØ╝ņ£╝Ēé©ļŗż. ņØ┤ļ¤¼ĒĢ£ ņāüĒśĖ ņ×æņÜ®ņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ļŖö ņ£äņäĀņĢö ļō▒ ņ¦łļ│æ ļ░£ņāØņØś ĻĖ░ņĀäņØä ņØ┤ĒĢ┤ĒĢśĻ│Ā ņ¦łļ│æ ļ░£ņāØņØä ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ļŖö ņ╣śļŻīļ▓ĢņØś Ļ░£ļ░£ņŚÉ ņ׳ņ¢┤ ļ¦żņÜ░ ņżæņÜöĒĢśļŗż. ņĢäņÜĖļ¤¼ ņÜ░ļ”¼ļéśļØ╝ņŚÉņä£ļÅä Ē¢źĒøä ĒÜ©Ļ│╝ņĀüņØĖ ņĀ£ĻĘĀņ╣śļŻīņÖĆ ņśłļ░® ļ░▒ņŗĀ Ļ░£ļ░£ņŚÉ ļģĖļĀźĒĢ£ļŗżļ®┤ H. pyloriņÖĆ Ļ┤ĆļĀ©ļÉ£ ņāüļČĆņ£äņןĻ┤Ć ņ¦łĒÖśņØś ņśłļ░®ņØĆ ļ¼╝ļĪĀ ņØ┤ņÖĆ Ļ┤ĆļĀ©ļÉ£ ņØśļŻīļ╣ä ņĀłĻ░ÉņŚÉļÅä Ēü¼Ļ▓ī ĻĖ░ņŚ¼ĒĢĀ Ļ▓āņ£╝ļĪ£ ĻĖ░ļīĆĒĢ£ļŗż.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






