


м„ң лЎ
нҸҗлҸҷл§Ҙ кі нҳҲм••мқҖ нҸҗмқҳ м„ёлҸҷл§Ҙ лӮҙн”јм„ёнҸ¬м—җ к·ёл¬јлӘЁм–‘лі‘н„°(plexiform lesion)лҘј мқјмңјмјң нҸҗлҸҷл§Ҙм••мқ„ мҰқк°ҖмӢңнӮҙмңјлЎңмҚЁ мҡ°мӢ¬л¶Җм „кіј н•Ёк»ҳ мӮ¬л§қмқ„ мҙҲлһҳн• мҲҳ мһҲлҠ” м§ҲнҷҳмқҙлӢӨ[1]. нҸҗлҸҷл§Ҙ кі нҳҲм••мқҳ м •мқҳлҠ” нҸҗм§Ҳнҷҳ, л§Ңм„ұнҳҲм „мғүм „м„ұнҸҗкі нҳҲм•• л“ұмқҙ мӣҗмқёмқҙ м•„лӢҢ нҸҗкі нҳҲм••мңјлЎңм„ң мӢ¬лҸ„мһҗлЎң мёЎм •н•ң нҸүк· нҸҗлҸҷл§Ҙм••мқҖ 25 mmHg мқҙмғҒ, нҸҗлӘЁм„ёнҳҲкҙҖ мҗҗкё°м••мқҖ 15 mmHg мқҙн•ҳмқҙлӢӨ. 1980л…„лҢҖ мҙҲ лҜёкөӯм—җм„ң 187лӘ…мқҳ нҠ№л°ңм„ұ нҸҗлҸҷл§Ҙкі нҳҲм•• нҷҳмһҗмқҳ м—ӯн•ҷ м—°кө¬м—җ мқҳн•ҳл©ҙ лӮЁм„ұліҙлӢӨ м—¬м„ұм—җм„ң нқ”н•ҳкі м§„лӢЁ лӢ№мӢң нҸүк· м—°л №мқҖ 35м„ёмқҙл©° м№ҳлЈҢн•ҳм§Җ м•ҠмқҖ кІҪмҡ° мӨ‘к°„ мғқмЎҙкё°к°„мқҖ 2.8л…„мқҙм—ҲлӢӨ[2]. нҸҗлҸҷл§Ҙ кі нҳҲм••мқҳ лі‘мқёлЎ мқҖ мһҳ л°қнҳҖм§Җм§Җ м•ҠмқҖ мғҒнғңмқҙм§Җл§Ң bone morphogenetic protein receptor type 2 (BMPR2) heterozygous лҸҢм—°ліҖмқҙк°Җ нҳ„мһ¬к№Ңм§Җ к°ҖмһҘ мһҳ м•Ңл Ө진 мң м „м Ғ мҶҢмқёмқҙлӢӨ. мқҙ BMPR2 лҸҢм—°ліҖмқҙлҠ” к°ҖмЎұм„ұ нҸҗлҸҷл§Ҙкі нҳҲм••мқҳ 70% мқҙмғҒ, нҠ№л°ңм„ұ нҸҗлҸҷл§Ҙкі нҳҲм••мқҳ 11-40%м—җм„ң л°ңкІ¬лҗңлӢӨ[3,4]. нҳ„мһ¬к№Ңм§Җ көӯлӮҙм—җм„ң BMPR2 лҸҢм—°ліҖмқҙк°Җ мҰқлӘ…лҗң л°”лҠ” м—Ҷм—Ҳмңјл©° ліё мҰқлЎҖлҠ” көӯлӮҙмқҳ мІ« ліҙкі мқҙлӢӨ.
мҰқ лЎҖ
нҷҳ мһҗ: лӮЁмһҗ, 43м„ё
мЈј мҶҢ: к°қнҳҲкіј мҡҙлҸҷ мӢң нҳёнқЎкіӨлһҖ(NYHA class III)
кіјкұ°л Ҙ л°Ҹ нҳ„лі‘л Ҙ: нҷҳмһҗлҠ” 2003л…„ нҸҗкІ°н•өмқ„ 진лӢЁл°ӣкі 6к°ңмӣ”к°„ н•ӯкІ°н•өм ң м№ҳлЈҢлҘј л§Ҳм№ң нӣ„ мҷ„м№ҳ нҢҗм •мқ„ л°ӣм•ҳлӢӨ. мқҙнӣ„ нҳёнқЎкё° мҰқмғҒ м—Ҷмқҙ м§ҖлӮҙлҚҳ мӨ‘ 2010л…„ 10мӣ” л‘җ м°ЁлЎҖ к°қнҳҲн•ҳм—¬ нғҖлі‘мӣҗм—җ л°©л¬ён•ҳмҳҖкі нқүл¶Җ м—‘мҠӨм„ кІҖмӮ¬м—җм„ң нҸҗлҸҷл§Ҙ нҒ¬кё° мҰқк°Җ мҶҢкІ¬мңјлЎң нқүл¶Җ м»ҙн“Ён„° лӢЁмёөмҙ¬мҳҒ, мӢ¬мҙҲмқҢнҢҢлҘј мӢңн–үн•ң нӣ„ нҸҗлҸҷл§Ҙ кі нҳҲм••мқҙ мқҳмӢ¬лҗҳм–ҙ ліёмӣҗ лӮҙмӣҗн•ҳмҳҖлӢӨ.
к°ҖмЎұл Ҙ: нҷҳмһҗмқҳ м–ҙлЁёлӢҲк°Җ м¶ңмӮ° нӣ„ мӢ¬мһҘмқҙ мўӢм§Җ м•ҠлӢӨлҠ” мқҙм•јкё°лҘј л“Өм—ҲмңјлӮҳ кІҖмӮ¬н•ҙ ліё м ҒмқҖ м—Ҷмңјл©° 65м„ём—җ мң л°©м•”мңјлЎң мӮ¬л§қн–ҲлӢӨ. м•„лІ„м§ҖлҠ” нҷҳмһҗк°Җ нғңм–ҙлӮ л¬ҙл ө м—°лқҪмқҙ лҒҠкІЁ нҷ•мқён• мҲҳ м—Ҷм—ҲлӢӨ. нҷҳмһҗмқҳ м—¬лҸҷмғқмқ„ 비лЎҜн•ҳм—¬ к·ё мҷё лӢӨлҘё к°ҖмЎұм—җм„ңлҠ” нҸҗлҸҷл§Ҙ кі нҳҲм••м—җ лҢҖн•ң к°ҖмЎұл ҘмқҖ м—Ҷм—ҲлӢӨ.
мӢ мІҙкІҖмӮ¬ мҶҢкІ¬: лӮҙмӣҗ лӢ№мӢң нҳҲм•• 135/85 mmHg, л§Ҙл°• 분лӢ№ 82нҡҢ, нҳёнқЎ 분лӢ№ 20нҡҢ, мІҙмҳЁ 36в„ғмҳҖкі , кёүм„ұ лі‘мғү м—Ҷмқҙ мқҳмӢқмқҖ лӘ…лЈҢн•ҳмҳҖлӢӨ. нқүл¶Җ 진찰м—җм„ң мҡ°мӢ¬мӢӨ мңөкё° мҶҢкІ¬мқҙ мһҲм—Ҳмңјл©° мІӯ진 мӢң P2 мқҢмқҳ н•ӯ진, мӮјмІЁнҢҗм—ӯлҘҳ мһЎмқҢмқҖ л“ӨлҰ¬м§Җ м•Ҡм•ҳкі лӘ©м •л§Ҙм••мқҖ м •мғҒмқҙм—Ҳмңјл©° к°„кІҪм •л§Ҙ л°ҳмӮ¬(hepatojugular reflex)к°Җ кҙҖм°°лҗҳм—ҲлӢӨ. нҸҗ мІӯ진 мӢң нҠ№мқҙ мҶҢкІ¬мқҖ м—Ҷм—Ҳмңјл©° м–‘ н•ҳм§Җмқҳ мҳӨлӘ©л¶Җмў…лҸ„ м—Ҷм—ҲлӢӨ.
кІҖмӮ¬ мҶҢкІ¬: л§җмҙҲнҳҲм•Ў кІҖмӮ¬м—җм„ң нҳҲмғүмҶҢ 15.9 g/dL, л°ұнҳҲкө¬ 9,700/ОјL, нҳҲмҶҢнҢҗ 160,000/ОјLлЎң лӘЁл‘җ м •мғҒмқҙм—ҲлӢӨ. лҸҷл§ҘнҳҲк°ҖмҠӨкІҖмӮ¬м—җм„ңлҠ” pH 7.40, PaO2 87.1 mmHg, PaCO2 32.8 mmHg, HCO3-20.4 mEq/L, мӮ°мҶҢнҸ¬нҷ”лҸ„ 97.0%мҳҖлӢӨ. нҳҲм•Ўнҷ”н•ҷ кІҖмӮ¬м—җм„ң к°„ л°Ҹ мӢ мһҘкё°лҠҘ мқҙмғҒмқҖ м—Ҷм—Ҳкі м „н•ҙм§ҲлҸ„ м •мғҒмқҙм—ҲлӢӨ. NT-proBNP мҲҳм№ҳк°Җ 125.2 pg/mLлЎң мҰқк°Җлҗҳм–ҙ мһҲм—Ҳкі 6분 кұ·кё° кІҖмӮ¬м—җм„ң 512 m кІ°кіјлҘј ліҙмҳҖлӢӨ.
лӢЁмҲңл°©мӮ¬м„ л°Ҹ мӢ¬м „лҸ„ мҶҢкІ¬: нқүл¶ҖлӢЁмҲңл°©мӮ¬м„ кІҖмӮ¬м—җм„ң мӢ¬мһҘ비лҢҖ л°Ҹ м–‘мёЎ нҸҗлҸҷл§Ҙмқҳ нҷ•мһҘ мҶҢкІ¬мқҙ мһҲм—ҲлӢӨ. мӢ¬м „лҸ„мғҒ лҸҷмңЁлҸҷмңјлЎң мҡ°мӢ¬мӢӨ비лҢҖк°Җ мһҲм—ҲлӢӨ.
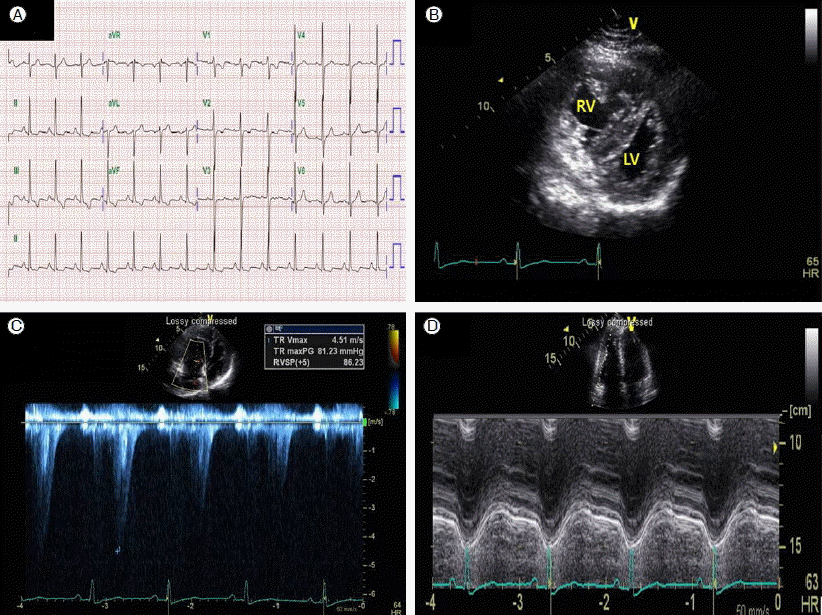
мӢ¬мһҘ л°Ҹ нҸҗ кІҖмӮ¬ мҶҢкІ¬: мӢ¬мҙҲмқҢнҢҢм—җм„ңлҠ” мўҢмӢ¬мӢӨмҲҳ축기м—җ мўҢмӢ¬мӢӨмӨ‘кІ©мқҙ лҲҢлҰ¬лҠ” DлӘЁм–‘ мўҢмӢ¬мӢӨ мҶҢкІ¬ л°Ҹ мҡ°мӢ¬мӢӨ 비нӣ„ мҶҢкІ¬мқҙ кҙҖм°°лҗҳм—Ҳмңјл©°, мӮјмІЁнҢҗл§үм—ӯлҘҳк°Җ м Ғм–ҙм„ң кіјмҶҢнҸүк°Җмқҳ к°ҖлҠҘм„ұмқҙ мһҲмқҢм—җлҸ„ л¶Ҳкө¬н•ҳкі нҸҗлҸҷл§Ҙм••л Ҙмқҙ 79 mmHgлЎң лҶ’кІҢ мёЎм •лҗҳм—ҲлӢӨ. м •мғҒ мўҢмӢ¬мӢӨл°•м¶ңкі„мҲҳ(59%), кІҪлҸ„мқҳ мҡ°мӢ¬мӢӨ нҷ•мһҘ л°Ҹ кІҪлҸ„мқҳ мҡ°мӢ¬мӢӨ кё°лҠҘ м Җн•ҳ(tricuspid annular plane systolic excursion = 19.5 mm)к°Җ кҙҖм°°лҗҳм—ҲлӢӨ(Fig. 1). мўҢмӢ¬мӢӨкё°лҠҘмқҖ м •мғҒмқҙм—ҲлӢӨ. нҸҗкё°лҠҘ кІҖмӮ¬м—җм„ң FEV1/FVC 72%, FVC 3.81 L (мҳҲмёЎм№ҳмқҳ 93%)лЎң м •мғҒ нҸҗкё°лҠҘмқ„ ліҙмҳҖмңјлӮҳ DLCo 64%лЎң кІҪлҸ„мқҳ нҷ•мӮ°лҠҘ м Җн•ҳлҘј ліҙмҳҖлӢӨ. нқүл¶Җ м „мӮ°нҷ”лӢЁмёөмҙ¬мҳҒм—җм„ң лҡңл ·н•ң нҸҗмғүм „мқҙлӮҳ кё°кҙҖм§ҖлҸҷл§Ҙмқҳ 비нӣ„лҠ” кҙҖм°°лҗҳм§Җ м•Ҡм•ҳмңјл©° мҡ°н•ҳм—Ҫм—җ кҙ‘лІ”мң„н•ң к°„мң лҰ¬ мқҢмҳҒмқҙ ліҙмҳҖлҠ”лҚ° мқҙлҠ” к°қнҳҲкіј кҙҖл Ён•ң нҸҗ лӮҙ м¶ңнҳҲмқ„ мӢңмӮ¬н•ҳмҳҖлӢӨ. мӢ¬лҸ„мһҗ кІҖмӮ¬м—җм„ң мҲҳ축기 нҸҗлҸҷл§Ҙм••л ҘмқҖ 100 mmHg, нҸүк· нҸҗлҸҷл§Ҙм••л ҘмқҖ 62 mmHg, нҸҗлӘЁм„ёнҳҲкҙҖнҸүк· м••мқҖ 6 mmHg, мӢ¬л°•м¶ңлҹү 4.7L/min, нҸҗлҸҷл§Ҙм Җн•ӯмқҖ 22.4 Wood unit/m2мңјлЎң мӨ‘мҰқмқҳ нҸҗлҸҷл§Ҙкі нҳҲм••м—җ н•©лӢ№н•ң мҶҢкІ¬мқҙм—ҲлӢӨ. мқјмӮ°нҷ”м§ҲмҶҢ нқЎмһ…мқ„ мқҙмҡ©н•ң кёүм„ұнҳҲкҙҖнҷ•мһҘкІҖмӮ¬м—җлҠ” л°ҳмқ‘мқҙ м—Ҷм—ҲлӢӨ.
мң м „мһҗ кІҖмӮ¬: BMPR2 мң м „мһҗ кІҖмӮ¬мғҒ exon 8мқҳ 1042-1047лІҲм§ё мҪ”лҸҲмқҳ кІ°мҶҗмңјлЎң 348лІҲм§ё м•„лҜёл…ёмӮ°мқё valineкіј 349лІҲм§ё м•„лҜёл…ёмӮ°мқё isoleucineмқҙ кІ°мҶҗлҗң мң м „мһҗ ліҖнҳ•(c.1042_1047del-GTTATT)мқҙ нҷ•мқёлҗҳм—ҲлӢӨ. мқҙлҠ” кё°мЎҙм—җ ліҙкі лҗҳм§Җ м•ҠмқҖ мғҲлЎңмҡҙ мң м „мһҗліҖнҳ•мқҙлӢӨ. нҷҳмһҗмқҳ к°ҖмЎұ мң м „мһҗ кІҖмӮ¬лҠ” мң мқјн•ң к°ҖмЎұмқё м—¬лҸҷмғқм—җкІҢм„ңл§Ң мӢңн–үлҗҳм—Ҳкі лҸҷмқј мң м „мһҗ ліҖнҳ•мқҖ кҙҖм°°лҗҳм§Җ м•Ҡм•ҳкё° л•Ңл¬ём—җ, м •мғҒ лҢҖмЎ°кө° н•ңкөӯмқё 50лӘ…мқ„ 분м„қн•ҳмҳҖлӢӨ. лҢҖмЎ°кө° 50лӘ… лӘЁл‘җм—җм„ң лҸҷмқј мң м „мһҗ ліҖнҳ•мқҖ кҙҖм°°лҗҳм§Җ м•Ҡм•„ c.1042_1047delGTTATTмқҙ ліё нҷҳмһҗм—җм„ң нҸҗлҸҷл§Ҙкі нҳҲм••мқ„ мқјмңјнӮЁ мӣҗмқё лҸҢм—°ліҖмқҙлЎң нҢҗлӢЁн•ҳмҳҖлӢӨ.
м№ҳлЈҢ л°Ҹ мһ„мғҒкІҪкіј: нҷҳмһҗлҠ” нҠ№л°ңм„ұ нҸҗлҸҷл§Ҙкі нҳҲм••мқ„ 진лӢЁл°ӣкі bosentan 62.5 mg 1мқј 2нҡҢ ліөмҡ©н•ҳкё° мӢңмһ‘н–Ҳкі лҸҷмӢңм—җ н•ӯмқ‘кі м ң м№ҳлЈҢлҘј лі‘н–үн•ҳмҳҖлӢӨ. м№ҳлЈҢ мӢңмһ‘ м „ 6분 ліҙн–үкІҖмӮ¬м—җм„ң 512 mлЎң мёЎм •лҗҳм—Ҳмңјл©° м№ҳлЈҢ к°ңмӢң 3к°ңмӣ” л’Ө 525 mлЎң мҰқк°Җн•ҳмҳҖлӢӨ. NT pro-BNPлҠ” 125 pg/mLм—җм„ң 40 pg/mLлЎң к°җмҶҢн•ҳмҳҖкі мҡҙлҸҷм„ұ нҳёнқЎкіӨлһҖмқҳ м •лҸ„к°Җ NYHA кё°лҠҘм„ұ 분лҘҳ IIIм—җм„ң IIлЎң нҳём „лҗҳм—ҲлӢӨ.
кі м°°
нҸҗлҸҷл§Ҙ кі нҳҲм••мқҖ 1) мӣҗмқёмқ„ м•Ң мҲҳ м—ҶлҠ” нҠ№л°ңм„ұ нҸҗлҸҷл§Ҙкі нҳҲм••, 2) мӣҗмқё мң м „мһҗк°Җ л°қнҳҖмЎҢкұ°лӮҳ к°ҖмЎұл Ҙмқҙ м•Ңл Ө진 мң м „м„ұ нҸҗлҸҷл§Ҙкі нҳҲм••, 3) мӢқмҡ•к°җнҮҙм ң л“ұмқҳ м•Ҫн’ҲмқҙлӮҳ лҸ…к·№л¬јм—җ л…ём¶ңлҗҳм–ҙ л°ңмғқн•ҳлҠ” кІҪмҡ°, 4) кё°м Җм§Ҳнҷҳкіј м—°кҙҖлҗң нҸҗлҸҷл§Ҙкі нҳҲм••(кІ°н•©мЎ°м§Ғлі‘, HIV к°җм—ј, л¬ёл§Ҙкі нҳҲм••, м„ мІңмӢ¬мһҘлі‘, мЈјнҳҲнқЎм¶©мҰқ[schistosomiasis], л§Ңм„ұ мҡ©нҳҲл№ҲнҳҲ)мңјлЎң кө¬л¶„лҗңлӢӨ[5]. нҸҗлҸҷл§Ҙ кі нҳҲм••мқҳ мӣҗмқё мң м „мһҗлЎңлҠ” transforming growth factor-ОІ (TGF-ОІ) superfamilyмқҳ мӢ нҳём „лӢ¬м—җ кҙҖм—¬н•ҳлҠ” лӢЁл°ұм§Ҳл“Өмқё BMPR2, activinlike kinase-type 1 (ALK1)кіј endoglin (ENG)мқҳ лҸҢм—°ліҖмқҙк°Җ ліҙкі лҗҳм—ҲлӢӨ. мқҙ мӨ‘ BMPR2 мң м „мһҗ лҸҢм—°ліҖмқҙлҠ” к°ҖмһҘ мң лӘ…н•ҳкі л№„көҗм Ғ нқ”нһҲ л°ңкІ¬лҗҳлҠ” ліҖмқҙлЎңм„ң, м „мІҙ нҸҗлҸҷл§Ҙ кі нҳҲм•• нҷҳмһҗмқҳ м•Ҫ 6% м •лҸ„лҘј м°Ём§Җн•ҳлҠ” мң м „м„ұ нҸҗлҸҷл§Ҙ кі нҳҲм•• нҷҳмһҗмқҳ м•Ҫ 70%м—җм„ң л°ңкІ¬лҗҳл©° нҠ№л°ңм„ұ нҸҗлҸҷл§Ҙкі нҳҲм•• нҷҳмһҗмқҳ м•Ҫ 10-40%м—җм„ң кҙҖм°°лҗңлӢӨ[2,4,6].
BMPR2 мң м „мһҗлҠ” мҙқ 13к°ңмқҳ exonмңјлЎң кө¬м„ұлҗҳм–ҙ мһҲмңјл©° м—¬кё°м„ң л§Ңл“Өм–ҙм§ҖлҠ” 1,038к°ңмқҳ м•„лҜёл…ёмӮ°мңјлЎң кө¬м„ұлҗң лӢЁл°ұм§ҲмқҖ лӢӨлҘё type II TGF-ОІ мҲҳмҡ©мІҙл“ӨмІҳлҹј мӢ нҳё нҺ©нғҖмқҙл“ң(signal peptide), лҰ¬к°„л“ң кІ°н•© лҸ„л©”мқё(ligand binding domain), single transmembrane domain, serine/threonine нӮӨлӮҳм•„м ң лҸ„л©”мқё л°Ҹ м„ёнҸ¬м§Ҳ лҸ„л©”мқё(cytoplasmic domain)мңјлЎң мқҙлЈЁм–ҙм ё мһҲлӢӨ. нҸҗлҸҷл§Ҙкі нҳҲм•• нҷҳмһҗмқҳ мң м „мһҗ кІҖмӮ¬ кІ°кіјк°Җ 축м ҒлҗЁм—җ л”°лқј м§ҖкёҲк№Ңм§Җ 298мў…лҘҳмқҳ лҸҢм—°ліҖмқҙк°Җ нҸҗлҸҷл§Ҙ кі нҳҲм•• нҷҳмһҗмқҳ BMPR2 мң м „мһҗ кІҖмӮ¬ кІ°кіј л°қнҳҖмЎҢлӢӨ[7]. мқҙл“Ө лҸҢм—°ліҖмқҙлҠ” м•„лҜёл…ёмӮ°мқ„ м „мӮ¬н•ҳлҠ” мҪ”лҸҲ(codon)мқҙ мң м „мһҗ м•”нҳён•ҙлҸ…(translation)мқ„ мў…кІ°н•ҳлҠ” мў…лЈҢ мҪ”лҸҲ(termination codon)мңјлЎң л°”лҖҢлҠ” nonsense mutation, лӢӨлҘё мў…лҘҳмқҳ м•„лҜёл…ёмӮ°мқ„ мҪ”л“ңн•ҳкІҢ лҗҳлҠ” missense mutation, н•ң к°ң лҳҗлҠ” л‘җ к°ңмқҳ лүҙнҒҙл ҲмҳӨнғҖмқҙл“ң(nucleotide) мӮҪмһ… л°Ҹ кІ°мҶҗм—җ мқҳн•ң frameshift mutation, мҠӨн”ҢлқјмқҙмӢұ кіјм •м—җ н•„мҡ”н•ң лүҙнҒҙл ҲмҳӨнғҖмқҙл“ңмқҳ лҸҢм—°ліҖмқҙлЎң мҠӨн”ҢлқјмқҙмӢұм—җ мқҙмғҒмқҙ мғқкё°лҠ” splice-site mutation, л„“мқҖ лІ”мң„м—җм„ң кІҢлҶҲ DNAмқҳ мӨ‘ліө лҳҗлҠ” кІ°мҶҗ(gene duplication/deletion mutation) л“ұмңјлЎң лӮҳлү мҲҳ мһҲлӢӨ. нҳ„мһ¬к№Ңм§Җ м•Ңл Ө진 298мў… лҸҢм—°ліҖмқҙмқҳ л№ҲлҸ„лҠ” nonsense mutation (85/298), frame-shift mutation (73/298), splice-site mutation (26/298), gene duplication/deletion mutation (19/298), missense mutation (95/298)мқҙлӢӨ[7]. лҸҢм—°ліҖмқҙлҠ” BMPR2 мң м „мһҗмқҳ лӘЁл“ exonм—җм„ң л°ңмғқн• мҲҳ мһҲлҠ”лҚ°, serine/threonine нӮӨлӮҳм•„м ң лҸ„л©”мқём—җм„ң к°ҖмһҘ л§ҺмқҖ лҸҢм—°ліҖмқҙк°Җ кҙҖм°°лҗҳкі лӢӨмқҢмңјлЎң лҰ¬к°„л“ң кІ°н•© лҸ„л©”мқёкіј м„ёнҸ¬м§Ҳ лҸ„л©”мқём—җм„ң л§ҺмқҖ лҸҢм—°ліҖмқҙк°Җ кҙҖм°°лҗңлӢӨ[6,7]. Missense mutationмқҖ л°”лҖҗ м•„лҜёл…ёмӮ°м—җ мқҳн•ҙ BMPR2 лӢЁл°ұм§Ҳмқҳ кё°лҠҘм—җ мқҙмғҒмқҙ мһҲлҠ” кІҪмҡ°м—җл§Ң BMPR2 мң м „мһҗмқҳ лҸҢм—°ліҖмқҙлЎң кө¬л¶„лҗңлӢӨ. Serine/threonine нӮӨлӮҳм•„м ң лҸ„л©”мқёкіј лҰ¬к°„л“ң кІ°н•© лҸ„л©”мқём—җ 80% мқҙмғҒмқҳ missense mutationмқҙ 집мӨ‘лҗҳм–ҙ мһҲлҠ” кІғмңјлЎң ліҙм•„ мқҙ л‘җ лҸ„л©”мқёмқҙ BMPR2 лӢЁл°ұм§Ҳмқҳ кё°лҠҘм—җ мӨ‘мҡ”н•ң м—ӯн• мқ„ н•ҳлҠ” кІғмқ„ м•Ң мҲҳ мһҲлӢӨ[4,6,7].
BMPR2 лҸҢм—°ліҖмқҙлҘј к°Җм§Җкі мһҲлҠ” нҷҳмһҗмқҳ нҳҲкҙҖм—җм„ңлҠ” BMPR2 л°ңнҳ„мқҙ нҳ„м ҖнһҲ к°җмҶҢлҗҳм–ҙ мһҲмңјл©°[8] мқҙлҹ¬н•ң BMPR2мқҳ к°җмҶҢлҠ” нҳҲкҙҖлӮҙн”јм„ёнҸ¬мқҳ м„ёнҸ¬мһҗл©ёмӮ¬(apoptosis)лҘј мҙү진н•ҳм—¬ лӘЁм„ёнҳҲкҙҖмқҙм „м„ёлҸҷл§Ҙмқ„ мһғкІҢ н•ҳкұ°лӮҳ мқҙлҹ¬н•ң м„ёнҸ¬мһҗл©ёмӮ¬м—җ м Җн•ӯн• мҲҳ мһҲлҠ” нҳҲкҙҖлӮҙн”јм„ёнҸ¬л“Өмқҳ л№„м •мғҒм Ғмқё мҰқмӢқмңјлЎң мқён•ң к·ёл¬јлӘЁм–‘лі‘н„°мқҳ нҳ•м„ұмқ„ мң лҸ„н•ҳлҠ” кІғмңјлЎң м¶”м •лҗңлӢӨ[9].
ліё мҰқлЎҖм—җм„ң л°ңкІ¬лҗң exon8мқҳ лҸҢм—°ліҖмқҙлҠ” BMPR2мқҳ л‘җ лҢҖлҰҪмң м „мһҗ(allele) мӨ‘ н•ң лҢҖлҰҪмң м „мһҗмқҳ нӮӨлӮҳм•„м ң лҸ„л©”мқёмқҳ 348, 349лІҲм§ё м•„лҜёл…ёмӮ° valineкіј isoleucineмқ„ мҪ”л“ңн•ҳлҠ” GTTATT м—јкё°м„ңм—ҙмқҙ кІ°мҶҗлҗҳм–ҙ л‘җ м•„лҜёл…ёмӮ°мқҙ м—Ҷм–ҙ진 BMPR2 лӢЁл°ұм§Ҳмқ„ л§Ңл“ӨкІҢ лҗҳлҠ” мғҲлЎңмҡҙ мң м „мһҗ ліҖнҳ•мқҙлӢӨ(Fig. 2) [10]. ліё м—°кө¬м§„мқҖ лӢӨмқҢмқҳ мқҙмң лЎң c.1042_1047delGTTATT ліҖмқҙк°Җ нҸҗлҸҷл§Ҙ кі нҳҲм••мқҳ мӣҗмқёмқё лҸҢм—°ліҖмқҙ мң м „мһҗлЎң кІ°лЎ мқ„ м§Җм—ҲлӢӨ. мІ«м§ё, кІ°мҶҗлҗң л‘җ м•„лҜёл…ёмӮ°мқҙ BMPR2 лӢЁл°ұм§Ҳм—җм„ң мӨ‘мҡ”н•ң м—ӯн• мқ„ н•ҳлҠ” нӮӨлӮҳм•„м ң лҸ„л©”мқём—җ мҶҚн•ңлӢӨ. л‘ҳм§ё, мқҙлҠ” м–ҙлҘҳм—җм„ңл¶Җн„° мӮ¬лһҢм—җ мқҙлҘҙкё°к№Ңм§Җ BMPR2 лӢЁл°ұм§Ҳм—җм„ң лҸҷмқјн•ҳкІҢ ліҙмЎҙлҗҳкі мһҲлҠ” м•„лҜёл…ёмӮ°л“ӨмқҙлӢӨ. м…Ӣм§ё, 348лІҲм§ё м•„лҜёл…ёмӮ°мқё valineмқҙ glutamic acidлЎң ліҖн•ң лҸҢм—°ліҖмқҙк°Җ кё°мЎҙмқҳ мң м „м„ұ нҸҗлҸҷл§Ҙ кі нҳҲм•• нҷҳмһҗмқҳ к°Җкі„м—җм„ң ліҙкі лҗң л°” мһҲлӢӨ[7]. л„·м§ё, ліё нҷҳмһҗмқҳ к°ҖмЎұ мӨ‘ нҸҗлҸҷл§Ҙ кі нҳҲм••мқ„ 진лӢЁл°ӣмқҖ к°ҖмЎұмқҖ м—Ҷм—ҲмңјлӮҳ нҸҗлҸҷл§Ҙкі нҳҲм••мқҙ мҰқлӘ…лҗҳм§Җ м•ҠмқҖ нҷҳмһҗмқҳ м—¬лҸҷмғқм—җ лҢҖн•ҙ BMPR2 мң м „мһҗмқҳ м§Ғм ‘ м—јкё°м„ңм—ҙмқ„ 분м„қн–ҲмңјлӮҳ нҷҳмһҗм—җм„ң л°ңкІ¬лҗң лҸҢм—°ліҖмқҙ(c.1042_1047-delGTTATT)лҠ” кҙҖм°°лҗҳм§Җ м•Ҡм•ҳлӢӨ. лӢӨм„Ҝм§ё, лҳҗн•ң м •мғҒ лҢҖмЎ°кө° н•ңкөӯмқё 50лӘ…мқҳ мң м „мһҗ 분м„қм—җм„ңлҸ„ лҸҷмқј лҸҢм—°ліҖмқҙлҠ” кҙҖм°°лҗҳм§Җ м•Ҡм•ҳлӢӨ. л”°лқјм„ң мғҒкё° м—јкё°м„ңм—ҙмқҳ ліҖнҷ”к°Җ ліё нҷҳмһҗм—җм„ң нҸҗлҸҷл§Ҙкі нҳҲм••мқ„ мқјмңјнӮЁ мӣҗмқё лҸҢм—°ліҖмқҙлЎң нҢҗлӢЁн•ҳмҳҖлӢӨ.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






