


ņä£ ļĪĀ
ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ņĘīņן ņÖĖļČäļ╣ä ņŗżņ¦łņØś ĒīīĻ┤┤ņÖĆ ņä¼ņ£ĀĒÖöļĪ£ ņØĖĒĢ£ ņĘīņן ņÖĖļČäļ╣ä ĻĖ░ļŖźņØś Ļ░Éņåī ļ░Å ĻČüĻĘ╣ņĀüņ£╝ļĪ£ ļé┤ļČäļ╣ä ņŗżņ¦łņØś ĒīīĻ┤┤Ļ╣īņ¦Ć ļÅÖļ░śĒĢśļŖö ļ╣äĻ░ĆņŚŁņĀüņØĖ ņĘīņןņØś ļ¦īņä▒ ņŚ╝ņ”Øņä▒ ņ¦łĒÖśņØ┤ļŗż[1]. ņØ┤ļ¤¼ĒĢ£ ļ│ĆĒÖöļĪ£ ņØĖĒĢśņŚ¼ ņ×¼ļ░£ņä▒ ļśÉļŖö ņ¦ĆņåŹņĀüņØĖ ļ│ĄĒåĄņØ┤ ņ£Āļ░£ļÉśĻ│Ā, ĒØĪņłśņןņĢĀ ļ░Å ļŗ╣ļć©Ļ░Ć ņ┤łļלļÉ£ļŗż. ņĪ░ņ¦ü ļ│æļ”¼ĒĢÖņĀüņ£╝ļĪ£ļŖö ļ╣äĻ░ĆņŚŁņĀü ĒĢ┤ļČĆĒĢÖņĀü ņĘīņןņØś ņåÉņāüņØä ņłśļ░śĒĢśļŖö ņŚ╝ņ”Øņä▒ ņ¦łĒÖśņØ┤ļ®░ ļ╣äĻ░ĆņŚŁņĀü ņĘīņןņåÉņāüņØĆ ņĘīņäĀļ░®ņäĖĒż(acinar cell)ņØś ņåīņŗż, ņä¼ņ£ĀĒÖö, tubular complexņØś ĒśĢņä▒ ļō▒ņØś ĒŖ╣ņä▒ņØä Ļ░¢ļŖöļŗż. ĻĖēņä▒ņĘīņןņŚ╝ņØĆ ņĘīņןĻĖ░ļŖźĻ│╝ ņĪ░ņ¦üĒĢÖņĀü ņåÉņāüņØ┤ ļīĆļČĆļČä ņĀĢņāüņ£╝ļĪ£ ĒÜīļ│ĄļÉśļéś ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ļŖö ņĘīņןņåÉņāüņØ┤ ņśüĻĄ¼ņĀüņ£╝ļĪ£ ļé©ĻĖ░ ļĢīļ¼ĖņŚÉ ņĀäĒåĄņĀüņ£╝ļĪ£ ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ļ│äĻ░£ņØś ņ¦łĒÖśņ£╝ļĪ£ ņŚ¼Ļ▓©ņĀĖ ņÖöļŗż. ĒĢśņ¦Ćļ¦ī ņ×äņāüņĀüņ£╝ļĪ£ ļ¦ÄņØĆ Ļ▓ĮņÜ░ ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØä ļ¬ģĒÖĢĒĢśĻ▓ī ĻĄ¼ļČäĒĢśĻĖ░ ņ¢┤ļĀżņÜ░ļ®░ ņŗżņĀ£ļĪ£ ņĢäņ¦üĻ╣īņ¦Ć ņĘīņןņŚ╝ņØś ļČäļźśņŚÉļÅä ļ¦ÄņØĆ Ēś╝ļ×ĆņØ┤ ņ׳ļŗż[1]. ņØ┤ļ¤¼ĒĢ£ ņ¦łļ│æ ļČäļźś ļ░Å ņĀĢņØśņØś Ēś╝ļ×ĆņØĆ ņĘīņןņØś ĒĢ┤ļČĆĒĢÖņĀü ņ£äņ╣śļĪ£ ņØĖĒĢśņŚ¼ ņĪ░ņ¦ü ņ¦äļŗ©ņØ┤ ļīĆļČĆļČä ņØ┤ļŻ©ņ¢┤ņ¦Ćņ¦Ć ņĢŖĻ│Ā ņ׳ņ£╝ļ®░ ņĘīņןņŚ╝ņØś ņĪ░ĻĖ░ ņśüņāüĒĢÖņĀü ņ¦äļŗ©ņØ┤ ļ¬©ĒśĖĒĢśĻ│Ā ļ░®ņé¼ņäĀ ņśüņāüĻ│╝ ņĪ░ņ¦ü ļ│ĆĒÖöņØś ņāüĻ┤ĆĻ┤ĆĻ│äĻ░Ć ļ░ØĒśĆņĀĖ ņ׳ņ¦Ć ņĢŖļŖöļŹ░ ĻĖ░ņØĖĒĢ£ļŗż. ĻĘĖļ¤¼ļéś ĻĖēņä▒ņĘīņןņŚ╝ņØ┤ Ļ│äņåŹĒĢ┤ņä£ ņ×¼ļ░£ĒĢśļ®┤ ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ ņ¦äĒ¢ēļÉĀ ņłś ņ׳Ļ│Ā, ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ļ│æ ņøÉņØĖņØ┤ ņāüļŗ╣ļČĆļČä Ļ▓╣ņ╣śĻ│Ā, Ļ░ÖņØĆ ņŗżĒŚśļ¬©ļŹĖņŚÉņä£ ĒöäļĪ£ĒåĀņĮ£ņØä ņłśņĀĢĒĢśļ®┤ Ļ░üĻ░üņØś ņāüĒā£Ļ░Ć ņ£Āļ░£ļÉĀ ņłś ņ׳ņ£╝ļ®░ ĻĖēņä▒ņŚ╝ņ”Øņŗ£ņØś ņ×äņāüņ¢æņāüĻ│╝ Ēśłņ▓ŁĒĢÖņĀü ļ│ĆĒÖöĻ░Ć ļ╣äņŖĘĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ Ēśäņ×¼ļŖö ĻĖēņä▒ņĘīņןņŚ╝, ņ×¼ļ░£ņä▒ ĻĖēņä▒ņĘīņןņŚ╝, ĻĘĖļ”¼Ļ│Ā ļ¦īņä▒ņĘīņןņŚ╝ņØä Ļ░ÖņØĆ ņ¦łļ│æņØś ņŚ░ņåŹņäĀņāüņŚÉ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ļ│┤ļŖö Ļ▓āņØ┤ ĒāĆļŗ╣ĒĢśļŗżĻ│Ā ņŚ¼Ļ▓©ņ¦ĆĻ│Ā ņ׳ļŗż[2-5].
ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æņØĖņ£╝ļĪ£ļŖö ļ¦īņä▒ ņĢīņĮöņś¼ ņäŁņĘ©, ļČĆĻ░æņāüņäĀĻĖ░ļŖźĒĢŁņ¦äņ”Ø, ļŗ┤ņäØ ļō▒ņØś ļŗ┤Ļ┤ĆĻ│ä ņ¦łĒÖś, ņÖĖņāü, ņĘīņןņä¼ņ£Āņóģ, ņĘīņןĻĖ░ĒśĢ ļō▒ņØ┤ ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░, ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś 30-40%ļŖö ĒŖ╣ļ│äĒĢ£ ņøÉņØĖņØä ļ░£Ļ▓¼ĒĢĀ ņłś ņŚåļŖö ĒŖ╣ļ░£ņä▒ņØĖ Ļ▓ĮņÜ░ņØ┤ļŗż[1]. Whitcomb ļō▒ņØĆ ļ¦īņä▒ņĘīņןņŚ╝ ļ░£ņāØņŚÉ Ļ┤ĆļĀ©ņØ┤ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉśļŖö ņøÉņØĖļōżņØś ņ▓½ ĻĖĆņ×Éļź╝ ļö░ņä£ TIGAR-O ļČäļźś(Toxic-metabolic, Idiopathic, Genetic, Autoimmune, Recurrent and severe acute pancreatitis associated chronic pancreatitis, Obstructive chronic pancreatitis)ļź╝ ņĀ£ņŗ£ĒĢśņśĆļŗż(Table 1) [6]. Ļ░ü ņøÉņØĖņŚÉ ņØśĒĢ£ ņĀĢĒÖĢĒĢ£ ļ░£ļ│æ ĻĖ░ņĀäņØĆ ņĢäņ¦ü ĻĘ£ļ¬ģļÉśņ¦Ć ņĢŖĻ│Ā ņ׳ņ¦Ćļ¦ī, ņĄ£ĻĘ╝ ņĘīņןņä¼ņ£ĀĒÖö ņāØņä▒ Ļ│╝ņĀĢņØś ņŚ░ĻĄ¼ņŚÉ ņ׳ņ¢┤ Ļ┤äļ¬®ĒĢĀ ļ¦īĒĢ£ ņ¦äļ│┤ņÖĆ ņ£ĀņĀäĒĢÖņĀü ļČäņ×É ņāØļ¼╝ĒĢÖņĀü ņŚ░ĻĄ¼ņØś ņ¦äņĀäņ£╝ļĪ£ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æņØĖļĪĀņØ┤ ņāłļĪŁĻ▓ī ņØĖņŗØļÉśĻ│Ā ņ׳ļŗż.
ļ│Ė Ļ│ĀņŚÉņä£ļŖö ļ©╝ņĀĆ TIGAR-O ļČäļźśņŚÉ ļö░ļźĖ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æņØĖļĪĀņŚÉ Ļ┤ĆĒĢśņŚ¼ ņĢīņĢäļ│┤Ļ│Ā, ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ ņä¼ņ£ĀĒÖöņØś ņ¦äĒ¢ēņŚÉ ļīĆĒĢ£ ņĄ£ņŗĀ ņ¦ĆĻ▓¼Ļ│╝ ļ│æņØĖļĪĀ ņżæņŚÉņä£ Ļ░Ćņן ĒØöĒĢ£ ņĢīņĮöņś¼ņä▒ ņĘīņןņŚ╝ņØś ļ░£ļ│æĻĖ░ņĀäņØä ņé┤ĒÄ┤ļ│Ė Ēøä ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝ņŚÉ ļīĆĒĢśņŚ¼ ĻĖ░ņłĀĒĢśĻ│Āņ×É ĒĢ£ļŗż.
ļ│Ė ļĪĀ
ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æņØĖļĪĀ(etiology of chronic pancreatitis)
Toxic-metabolic
ņĢīņĮöņś¼(alcohol)
ņĢīņĮöņś¼ņØĆ ņśżļ×½ļÅÖņĢł ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ░Ćņן ĒØöĒĢ£ ņøÉņØĖņ£╝ļĪ£ ņĢīļĀżņĀĖ ņÖöĻ│Ā ņØ┤ļŖö ņä£ņ¢æļ┐Éļ¦ī ņĢäļŗłļØ╝ ĒśĖņŻ╝, ĻĘĖļ”¼Ļ│Ā ņÜ░ļ”¼ļéśļØ╝ņÖĆ ņØ╝ļ│ĖņØä ĒżĒĢ©ĒĢ£ ļÅÖņ¢æņØś ņŚ¼ļ¤¼ ļéśļØ╝ņŚÉņä£ļÅä ņŻ╝ņÜö ļ░£ļ│æ ņøÉņØĖņØ┤ļŗż[7]. ņĄ£ĻĘ╝ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ Ļ┤ĆĒĢśņŚ¼ ņØ┤Ēā£ļ”¼ņŚÉņä£ ņŗ£Ē¢ēĒĢ£ ļŗżĻĖ░Ļ┤Ć ņäżļ¼ĖņĪ░ņé¼ņŚÉ ņØśĒĢśļ®┤ ņĢīņĮöņś¼ņØ┤ ņøÉņØĖņØ┤ņŚłļŹś Ļ▓ĮņÜ░Ļ░Ć 34% ļ░¢ņŚÉ ļÉśņ¦Ć ņĢŖņĢśĻ│Ā[8], ļ»ĖĻĄŁņŚÉņä£ ņŗ£Ē¢ēļÉ£ North American Pancreatitis Study 2 (NAPS2)ņŚÉņä£ļÅä ņĢīņĮöņś¼ņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņøÉņØĖņØ┤ņŚłļŹś Ļ▓ĮņÜ░ļŖö 45%ļĪ£ ļé«ņØĆ ļ╣äņ£©ņØä ņ░©ņ¦ĆĒĢśĻĖ┤ Ē¢łņ¦Ćļ¦ī[9], ņĢäņ¦üĻ╣īņ¦ĆļÅä ņä£ĻĄ¼ņŚÉņä£ ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ░Ćņן ĒØöĒĢ£ ņøÉņØĖņØ┤ ņĢīņĮöņś¼ņØ┤ļØ╝ļŖö ņĀÉņŚÉ ļīĆĒĢ┤ņä£ ņØ┤Ļ▓¼ņØ┤ ņŚåļŗż. ņĢīņĮöņś¼ļĪ£ ņØĖĒĢ£ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ļīĆĻ░£ ĒĢśļŻ©ņŚÉ 150 gņØś ņĢīņĮöņś¼(ņåīņŻ╝ 2ļ│æ)ņØä 10-15ļģä ļÅÖņĢł ļ¦żņØ╝ ļ│ĄņÜ®ĒĢśļŖö Ļ▓ĮņÜ░ ļ░£ņāØĒĢĀ ņłś ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļéś[10], ņĢīņĮöņś¼ņŚÉ ņØśĒĢ£ ļ¦īņä▒ņĘīņןņŚ╝ ļ░£ņāØņŚÉ ļīĆĒĢ£ ļ│æĒā£ ņāØļ”¼ĒĢÖņĀü ņØ┤ĒĢ┤ļŖö ļ¦żņÜ░ ļČĆņĪ▒ĒĢ£ ņŗżņĀĢņØ┤ļŗż. ņØ┤ļŖö ļ¦īņä▒ņØīņŻ╝ņ×ÉņØś 10% ļ»Ėļ¦īņŚÉņä£ ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ ļ░£ņāØĒĢśļ®░ ņĢīņĮöņś¼ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņĢĮ 30%ņØś Ļ▓ĮņÜ░ļŖö ņĢīņĮöņś¼ņØś ņäŁņĘ©ļ¤ēņØ┤ ļ╣äĻĄÉņĀü Ļ▓Įļ»ĖĒĢśņŚ¼ ņøÉņØĖ Ļ▓░Ļ│╝ņØś ņāüĻ┤ĆĻ┤ĆĻ│äĻ░Ć ļåÆņ¦Ć ņĢŖņĢä ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ņāØņŚÉ ņĢīņĮöņś¼ ņØ┤ņÖĖ ĒÖśĻ▓Įņ£ĀņĀäĒĢÖņĀü ņÜöņØĖņØ┤ ņ×æņÜ®ĒĢĀ Ļ▓āņØ┤ļØ╝ļŖö ņé¼ņŗżĻ│╝ ĒĢ©Ļ╗ś ņ×äņāüņĀüņ£╝ļĪ£ ĒĢ®ļŗ╣ĒĢ£ ņŗżĒŚśņĀü ņĢīņĮöņś¼ņĘīņןņŚ╝ ļÅÖļ¼╝ļ¬©ļŹĖņØ┤ ņŚåļŗżļŖö ņé¼ņŗżņŚÉ ĻĖ░ņØĖĒĢ£ļŗż[11-13].
ĒØĪņŚ░(tobacco smoking)
ĒØĪņŚ░Ļ│╝ ĻĖēņä▒ņĘīņןņŚ╝ ĻĘĖļ”¼Ļ│Ā ļ¦īņä▒ņĘīņןņŚ╝ņØś ņŚ░Ļ┤Ćņä▒ņŚÉ ļīĆĒĢ┤ņä£ļŖö ņŚ¼ļ¤¼ ņ░©ļĪĆ ļ│┤Ļ│ĀĻ░Ć ļÉśņŚłĻ│Ā Ēśäņ×¼ ĒØĪņŚ░ņØĆ ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ņØś ņ¦äĒ¢ēņØä ņ┤ēņ¦äĒĢśļŖö ņ¢æņØś ņāüĻ┤ĆĻ┤ĆĻ│ä(dose-dependent)ļź╝ ļ│┤ņØ┤ļŖö ļÅģļ”ĮņĀüņØĖ ņ£äĒŚśņØĖņ×ÉļĪ£ ņŚ¼Ļ▓©ņ¦ĆĻ│Ā ņ׳ļŗż[9,11,14]. ĻĘĖ ĻĖ░ņĀäņØĆ ņĢäņ¦ü ĻĘ£ļ¬ģļÉśņ¦Ć ņĢŖņĢśņ£╝ļéś ĒØĪņŚ░ ņŗ£ ņĘīņןņØś ņżæĒāäņé░ņŚ╝ ļČäļ╣äņØś ņĀĆĒĢśņÖĆ Ēśłņżæ ĒŖĖļ”ĮņŗĀ ņ¢ĄņĀ£ļŖź, ĻĘĖļ”¼Ļ│Ā ╬▒1-antitrypsinņ╣śņØś Ļ░ÉņåīĻ░Ć Ļ┤ĆņŚ¼ĒĢĀ Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉśĻ│Ā ņ׳ļŗż.
Idiopathic
ņĢīņĮöņś¼ņØ┤ ņøÉņØĖņØ┤ ņĢäļŗī ļ¦īņä▒ņĘīņןņŚ╝ņØś ņāüļŗ╣ņłś(30-40%)ļŖö ĒŖ╣ļ░£ņä▒(idiopathic) ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ ļČäļźśļÉ£ļŗż[1]. ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ļīĆĻ░£ņØś Ļ▓ĮņÜ░ ņøÉņØĖņØ┤ ļ░ØĒśĆņ¦Ćņ¦ĆļŖö ņĢŖņ£╝ļéś ņØ╝ļČĆļŖö ņĢīņĮöņś¼ņŚÉ ļīĆĒĢ£ Ļ│╝ļ»╝ļ░śņØæ, ļ¬©ļź┤Ļ│Ā ņ¦ĆļéśĻ░ĆĻ▒░ļéś ļ│┤Ļ│ĀļÉśņ¦Ć ņĢŖņØĆ ņĘīņןņÖĖņāü, ņ£ĀņĀäņĀü ņÜöņØĖ ļō▒ņØ┤ ņøÉņØĖņØ╝ Ļ░ĆļŖźņä▒ņØ┤ ņ׳ļŗż. ņØĖļÅäņÖĆ ņżæĻĄŁņØś Ļ▓ĮņÜ░ņŚÉļŖö ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś 60-70%, ņÜ░ļ”¼ļéśļØ╝ļŖö 20-59%, ĻĘĖļ”¼Ļ│Ā ņØ╝ļ│ĖņØĆ 35% ņĀĢļÅäĻ░Ć ĒŖ╣ļ░£ņä▒ņØĖ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[7,15]. ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝ņØĆ ņ▓ŁņåīļģäĒśĢ(juvenile ļśÉļŖö early onset)Ļ│╝ ļģĖņØĖĒśĢ(senile ļśÉļŖö late onset)ņ£╝ļĪ£ ļéśļēśļŖöļŹ░, ņ▓ŁņåīļģäĒśĢņØĆ 20ņäĖ ņĀäĒøäņŚÉ ĒśĖļ░£ĒĢśļ®░ ļÅÖĒåĄņØ┤ ņŗ¼ĒĢśņ¦Ćļ¦ī ņÖĖļČäļ╣ä ĻĖ░ļŖźņØ┤ ņóŗĻ│Ā ņäØĒÜīĒÖöļÅä ņŗ¼ĒĢśņ¦Ć ņĢŖņØĆ ļ░śļ®┤ ļģĖņØĖĒśĢņØĆ 60ņäĖ ņĀäĒøä(ļīĆĻ░£ļŖö 50-70ņäĖ)ņŚÉ ņל ļ░£ņāØĒĢśĻ│Ā ļÅÖĒåĄņØĆ ņŗ¼ĒĢśņ¦Ć ņĢŖņ£╝ļéś ņäØĒÜīĒÖöĻ░Ć ņŗ¼ĒĢśļŗż[16,17].
ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝(tropical pancreatitis)ņØĆ ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝ņØś ĒĢ£ ĒśĢĒā£ļĪ£ ņŻ╝ļĪ£ ņĀŖņØĆ ņé¼ļ×īņŚÉĻ▓ī ļ░£ņāØĒĢśĻ│Ā ļŗ╣ļć©ņØś ĒśĖļ░£Ļ│╝ ļæÉļō£ļ¤¼ņ¦ä ņĘīņןņØś ņäØĒÜīĒÖöĻ░Ć ĒŖ╣ņ¦ĢņĀüņØ┤ļŗż[18]. ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝ņØĆ ņśüņ¢æņŗżņĪ░Ļ░Ć ņŗ¼Ļ░üĒĢśĻ│Ā ņ╣┤ņé¼ļ░ö(cassava)ļØ╝ļŖö ņŗØņÜ® ļ┐īļ”¼ļź╝ ņŻ╝ņŗØņ£╝ļĪ£ ĒĢśļŖö ļé©ļČĆ ņØĖļÅäļź╝ ĒżĒĢ©ĒĢ£ ņĢäņŗ£ņĢä, ņĢäĒöäļ”¼ņ╣┤, ĻĘĖļ”¼Ļ│Ā ņżæņĢÖ ņĢäļ®öļ”¼ņ╣┤ ļō▒ņØś ņŚ┤ļīĆ ņ¦ĆņŚŁņŚÉ ņ£äņ╣śĒĢ£ Ļ░£ļ░£ļÅäņāüĻĄŁņŚÉņä£ ņŻ╝ļĪ£ ļ░£ņāØĒĢśĻĖ░ ļĢīļ¼ĖņŚÉ ņ╣┤ņé¼ļ░öĻ░Ć ĻĘĖ ņøÉņØĖņ£╝ļĪ£ ņāØĻ░üļÉśņŚłņŚłļŗż. ĻĘĖļ¤¼ļéś ņ╣┤ņé¼ļ░öĻ░Ć ļ¦īņä▒ņĘīņןņŚ╝ņØä ņØ╝ņ£╝Ēé©ļŗżļŖö ņŗżĒŚśņĀü ļśÉļŖö ņ×äņāüņĀü ņ”ØĻ▒░ļŖö ņŚåņ£╝ļ®░, Ļ▓īļŗżĻ░Ć ļé©ļČĆ ņØĖļÅäņŚÉņä£ ņ╣┤ņé¼ļ░öņØś ņäŁņĘ©Ļ░Ć ņżäņ¢┤ļōżņ¦Ć ņĢŖņĢśņØīņŚÉļÅä ļČłĻĄ¼ĒĢśĻ│Ā ņŗ£Ļ░äņØ┤ ņ¦Ćļé©ņŚÉ ļö░ļØ╝ ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝ņØ┤ Ļ│äņåŹĒĢ┤ņä£ Ļ░ÉņåīĒĢśņśĆļŗżļŖö Ļ▓āņØĆ ņ╣┤ņé¼ļ░öĻ░Ć ņøÉņØĖņØ┤ļØ╝Ļ│Ā ļ│┤ļŖöļŹ░ ļ¼┤ļ”¼Ļ░Ć ņ׳ņØīņØä ņŗ£ņé¼ĒĢ£ļŗż[19]. Ļ│ĄĻĄÉļĪŁĻ▓īļÅä ņØ┤ ņ¦ĆņŚŁņŚÉņä£ ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝ņØś Ļ░ÉņåīļŖö ļō▒ņ£Ā(kerosene) ļīĆņŗĀņŚÉ ņĀäĻĖ░ņØś ņé¼ņÜ®ņØ┤ ņ”ØĻ░ĆĒĢ£ Ļ▓āĻ│╝ ļ¦×ļ¼╝ļĀż ņ׳ĻĖ░ņŚÉ ļō▒ņ£ĀņŚÉņä£ ļéśņś© ļÅģņä▒ļ¼╝ņ¦łņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØä ņØ╝ņ£╝ņ╝░ļŗżĻ│Ā ļ│┤ļŖö Ļ▓āņØ┤ ļŹö ņäżļōØļĀźņØ┤ ņ׳ļŗżĻ│Ā ļ│┤Ļ│Ā ņ׳ļŗż. Ēśäņ×¼ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņøÉņØĖņ£╝ļĪ£ ņØĖļÅäņŚÉņä£ ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝ņØ┤ ņ░©ņ¦ĆĒĢśļŖö ļ╣äņ£©ņØĆ 3.8%ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[20]. ņ░ĖĻ│ĀļĪ£ ņŚ┤ļīĆņä▒ ņĘīņןņŚ╝ ĒÖśņ×ÉņØś 20-55%ņŚÉņä£ SPINK1 (serine protease inhibitor, Kazal type 1) ņ£ĀņĀäņ×É ļ│ĆņØ┤Ļ░Ć Ļ┤Ćņ░░ļÉśĻ│Ā CFTR (cystic fibrosis transmembrane conductance regulator) ņ£ĀņĀäņ×É ļ│ĆņØ┤ļÅä 50% ņĀĢļÅäņŚÉņä£ Ļ┤Ćņ░░ļÉśņ¢┤ ņä£ĻĄ¼ņØś ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ ļ│┤ņØ┤ļŖö ņ£ĀņĀäņ×É ļ│ĆņØ┤ ņåīĻ▓¼Ļ│╝ ļ╣äņŖĘĒĢśļŗżļŖö Ļ▓āņØĆ ĒØźļ»ĖļĪ£ņÜ┤ ņØ╝ņØ┤ļŗż[21].
Genetic
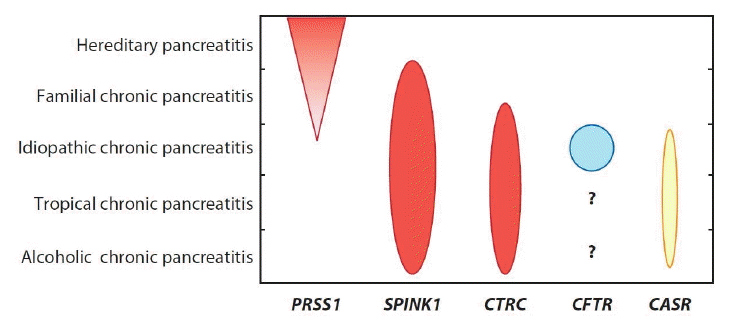
ņ£ĀņĀäņä▒ ņĘīņןņŚ╝(hereditary pancreatitis)ņØĆ 1952ļģä Comfort ļō▒ņŚÉ ņØśĒĢ┤ņä£ ņāüņŚ╝ņāēņ▓┤ ņÜ░ņä▒(autosomal dominant)ņ£╝ļĪ£ ņ£ĀņĀäļÉśļŖöņ¦łĒÖśņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłĻ│Ā 80% ņĀĢļÅäņØś ļ░£Ēśäņ£©ņØä ļéśĒāĆļéĖļŗż[22].ņ£ĀņĀäņä▒ ņĘīņןņŚ╝ņØĆ ļÅÖņØ╝ Ļ░ĆĻ│ä ļé┤ņŚÉņä£ ĻĖēņä▒ ļ░śļ│Ąņä▒ ļśÉļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ 2ņäĖļīĆ ņØ┤ņāüņŚÉ Ļ▒Ėņ│Éņä£ 3ļ¬ģ ņØ┤ņāüņØ┤ ņ׳ļŖö Ļ▓ĮņÜ░ ņ¦äļŗ©ļÉĀ ņłś ņ׳ļŖöļŹ░, ņ”ØņāüņØĆ ļīĆļČĆļČä 20ņäĖ ņØ┤ĒĢśņŚÉņä£ ņŗ£ņ×æļÉ£ļŗż. ņØ┤ļ¤¼ĒĢ£ ņĘīņןņŚ╝ņØś Ļ░ĆņĪ▒ļĀźņØĆ ņ׳ņ£╝ļéś ņĢ×ņŚÉņä£ ņ¢ĖĻĖēĒĢ£ ņ¦äļŗ© ĻĖ░ņżĆņØä ļ¦īņĪ▒ņŗ£Ēéżņ¦Ć ļ¬╗ĒĢśļŖö Ļ▓ĮņÜ░ Ļ░ĆņĪ▒ņä▒ ņĘīņןņŚ╝(familial pancreatitis)ņØ┤ļØ╝Ļ│Ā ļČĆļźĖļŗż. ņ£ĀņĀäņä▒ ņĘīņןņŚ╝ņØś ņøÉņØĖ ņ£ĀņĀäņ×É ļ│ĆņØ┤Ļ░Ć 1996ļģä ņ▓śņØī ļ░£Ļ▓¼ļÉ£ ņØ┤ļל ņłśĻ░£ņØś cationic trypsinogen (PRSS1; protease, serine, 1) ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤Ļ░Ć ļ░£Ļ▓¼ļÉśņŚłļŗż. ļśÉĒĢ£ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ CFTR ņ£ĀņĀäņ×ÉņØś ļÅīņŚ░ļ│ĆņØ┤ņÖĆ SPINK1 ņ£ĀņĀäņ×ÉņØś ņØ┤ņāüņØ┤ ļŗżņłś ļ░£Ļ▓¼ļÉśņŚłĻ│Ā, ņØ┤ņ¢┤ņä£ calcium-sensing receptor (CASR), anionic trypsinogen (PRSS2; protease, serine, 2), ĻĘĖļ”¼Ļ│Ā chymotrypsinogen C (CTRC) ņ£ĀņĀäņ×ÉļÅä ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ņāØ ĻĖ░ņĀäņŚÉ Ļ┤ĆņŚ¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņØś ņŚŁĒĢĀņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć Ļ│äņåŹĒĢ┤ņä£ ĒÖ£ļ░£Ē׳ ņØ┤ļŻ©ņ¢┤ņ¦ĆĻ│Ā ņ׳ļŗż(Table 2) (Fig. 1) [23].
PRSS1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤
1996ļģä Whitcomb ļō▒ņØĆ ņ£ĀņĀäņä▒ ņĘīņןņŚ╝ Ļ░ĆĻ│äņŚÉņä£ cationic trypsinogenņØä ļ¦īļōżņ¢┤ļé┤ļŖö PRSS1 ņ£ĀņĀäņ×ÉņŚÉ ļ│ĆņØ┤Ļ░Ć ņØ╝ņ¢┤ļéśņä£ ņĘīņןņŚ╝ņØ┤ ļ░£ņāØĒĢ£ļŗżļŖö Ļ▓āņØä ļ░ØĒśĆļāłļŗż[24,25]. ņ▓½ ļ▓łņ¦Ė ļ░£Ļ▓¼ļÉ£ ļ│ĆņØ┤ļŖö codon 122ņŚÉņä£ arginine (Arg, R)ņØ┤ histidine (His, H)ņ£╝ļĪ£ ņ╣śĒÖśļÉ£ R122H ļ│ĆņØ┤ņØ┤ļ®░, ņØ┤Ēøä N29I, A16V, K23R, N29T, R122C, D19A, D22G ļ│ĆņØ┤ ļō▒ņØ┤ ļ│┤Ļ│ĀļÉśņŚłļŗż.
ņĀĢņāüņĀüņ£╝ļĪ£ļŖö ĒŖĖļ”Įņŗ£ļģĖĻ▓É(trypsinogen)ņØ┤ ņĘīņןņŚÉņä£ ļ¦īļōżņ¢┤ņĀĖ ņŗŁņØ┤ņ¦Ćņןņ£╝ļĪ£ ļČäļ╣äļÉ£ ņØ┤ĒøäņŚÉņĢ╝ ĒŖĖļ”ĮņŗĀ(trypsin)ņ£╝ļĪ£ ļ│ĆĒÖśļÉśņ¢┤ ĒÖ£ņä▒ĒÖöļÉ£ļŗż. ļ¦īņĢĮ ņĘīņäĀļ░® ņäĖĒż ļé┤ņŚÉņä£ ĒŖĖļ”Įņŗ£ļģĖĻ▓ÉņØ┤ ļ╣äņĀĢņāüņĀüņ£╝ļĪ£ ĒÖ£ņä▒ĒÖöļÉśņ¢┤ ĒŖĖļ”ĮņŗĀņ£╝ļĪ£ ļ░öļĆīļ®┤ ņØ┤ņŚÉ ļīĆĒĢ£ ļ░®ņ¢┤ĻĖ░ņĀäņ£╝ļĪ£ ĒŖĖļ”ĮņŗĀņØ┤ ņŖżņŖżļĪ£ ļŗżļźĖ ĒŖĖļ”ĮņŗĀņØä ļČäĒĢ┤ņŗ£ņ╝£ ļ╣äĒÖ£ņä▒ĒÖö ĒĢ£ļŗż. ĒĢśņ¦Ćļ¦ī cationic trypsinogen ņ£ĀņĀäņ×ÉņØś ļīĆĒæ£ņĀüņØĖ ĻĖ░ļŖź ĒÜŹļōØņä▒ ļ│ĆņØ┤(gain-of-function mutation)ņØĖ R122Hļ│ĆņØ┤Ļ░Ć ļ░£ņāØĒĢśļ®┤ ĒŖĖļ”ĮņŗĀņŚÉ ņØśĒĢ┤ ņØĖņ¦ĆļÉśļŖö ļČĆņ£äņØś ĻĄ¼ņĪ░Ļ░Ć ļ│ĆĻ▓ĮļÉśņ¢┤ ĒŖĖļ”ĮņŗĀņŚÉ ņØśĒĢ£ ļČäĒĢ┤ņŚÉ ņĀĆĒĢŁņä▒ņØä Ļ░¢ļŖö ļ╣äņĀĢņāüņĀüņØĖ ĒŖĖļ”ĮņŗĀņØ┤ ĒśĢņä▒ļÉ£ļŗż. ĻĘĖļ”¼ĒĢśņŚ¼ ņØ┤ ļ╣äņĀĢņāüņĀüņØĖ ĒŖĖļ”ĮņŗĀņØ┤ ĒĢ£ļ▓ł ĒÖ£ņä▒ĒÖöĻ░Ć ļÉśļ®┤ ļČäĒĢ┤Ļ░Ć ļÉśņ¦Ć ņĢŖĻĖ░ ļĢīļ¼ĖņŚÉ Ļ│äņåŹĒĢ┤ņä£ ņĘīņןņŚ╝ņØä ņ£Āļ░£ĒĢśĻ▓ī ļÉ£ļŗż[24-26]. ļŗżņ¢æĒĢ£ ņ£ĀņĀäņ×ÉļōżņØ┤ ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņøÉņØĖņ£╝ļĪ£ ņØĖĒĢ£ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ļŗżņåī Ļ┤ĆļĀ©ņØ┤ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļéś PRSS1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņØś Ļ▓ĮņÜ░ņŚÉļŖö ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ļéś ņŚ┤ļīĆņä▒ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ņŚ░Ļ┤ĆņØ┤ ņŚåļŗżĻ│Ā ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[23].
CFTR ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤
ļéŁņä▒ņä¼ņ£Āņ”Ø(cystic fibrosis)ņØĆ CFTR ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņŚÉ ņØśĒĢ£ ņāüņŚ╝ņāēņ▓┤ ņŚ┤ņä▒ ņ£ĀņĀäņ¦łĒÖśņ£╝ļĪ£ ņĘīņןņŚÉņä£ļŖö ņĘīĻ┤ĆņäĖĒż(ductular cell)ņÖĆ ņĘīņäĀļ░®ņäĖĒżņØś ļČäļ╣ä ņןņĢĀļź╝ ņØ╝ņ£╝ņ╝£ ņ¦ĆņåŹņĀüņØĖ ņåÉņāüņØä ņ£Āļ░£ĒĢ£ļŗż[27]. ļéŁņä▒ņä¼ņ£Āņ”ØņØä ņØ╝ņ£╝ĒéżļŖö CFTR ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ļŖö ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉĻ▓īņä£ļÅä ļ░£Ļ▓¼ņØ┤ ļÉśļŖöļŹ░, ņØ┤ļōżņØĆ ņ×äņāüņĀüņ£╝ļĪ£ ņĀäĒśĢņĀüņØĖ ļéŁņä▒ņä¼ņ£Āņ”ØņØ┤ ņŚåļŖö Ļ▓ĮņÜ░ļÅä ļ¦ÄņĢśļŗż[28,29]. CFTR ņ£ĀņĀäņ×ÉļŖö cAMPņŚÉ ņØśĒĢ┤ ĒÖ£ņä▒ĒÖöļÉśļŖö ņāüĒö╝ņäĖĒż Ēæ£ļ®┤ņØś ļéśĒŖĖļź©-ņŚ╝ņåī ņłśņåĪļŗ©ļ░▒ņ¦łņØä ļ¦īļōżĻ│Ā ņŚ¼ļ¤¼ ņłśņåĪļŗ©ļ░▒ņ¦łņØś ņĪ░ņĀłņŚÉ Ļ┤ĆņŚ¼ĒĢ£ļŗż. ņĘīņןņŚÉņä£ļŖö cAMP ļ¦żĻ░£ņŚÉ ņØśĒĢ£ ņżæĒāäņé░ņŚ╝ņØś ļČäļ╣äļź╝ ņĪ░ņĀłĒĢśļŖöļŹ░, ņØ┤Ļ▓āņØś ņŻ╝ņÜö ĻĖ░ļŖźņØĆ ļŗ©ļ░▒ņ¦łņØ┤ ĒÆŹļČĆĒĢ£ ņĘīņä▒ļ░®ņäĖĒżņØś ļČäļ╣äļ¼╝ņØä ĒؼņäØņŗ£ĒéżĻ│Ā ņĢīņ╣╝ļ”¼ĒÖöĒĢśņŚ¼ ņĘīņןņŚÉ ņåÉņāüņØä ņżä ņłś ņ׳ļŖö ļŗ©ļ░▒ņĀä(protein plug)ņØś ĒśĢņä▒ņØä ņśłļ░®ĒĢśļŖö Ļ▓āņØ┤ļŗż[23]. ņĀĢņāüņĀüņ£╝ļĪ£ CFTRņØĆ ņĘīņäĀļ░®ņäĖĒżņŚÉņä£ Ļ┤ĆĻ░Ģļé┤ļĪ£ Ē¢źĒĢśļŖö Ēæ£ļ®┤ņŚÉ ņĪ┤ņ×¼ĒĢśļéś ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ļŖö ļ╣äņĀĢņāüņĀüņ£╝ļĪ£ ņäĖĒżņ¦ł ļé┤ņŚÉ ņ£äņ╣śĒĢśĻ▓ī ļÉ£ļŗż. ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņŚÉņä£ļŖö ņØ┤ļ¤¼ĒĢ£ CFTRņØś ļ╣äņĀĢņāüņĀüņØĖ ļČäĒżĻ░Ć ņŖżĒģīļĪ£ņØ┤ļō£ņŚÉ ņØśĒĢ┤ ņĀĢņāüĒÖöļÉśņ¢┤ Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ ņŚ╝ņ”ØņØ┤ Ļ░ÉņåīĒĢśĻ│Ā, ņĘīņןņØś ļČäļ╣äĻĖ░ļŖźņØ┤ ĒÜīļ│ĄļÉśļ®░, ņĘīņäĀļ░®ņäĖĒżĻ░Ć ņ×¼ņāØļÉśļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņĪīļŗż[30].
SPINK1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤
SPINK1ņØĆ pancreatic secretory trypsin inhibitor (PSTI)ļĪ£ļÅä ļČłļ”¼ļ®░, ņĘīņן ļé┤ņŚÉņä£ ĒŖĖļ”Įņŗ£ļģĖĻ▓ÉņØ┤ ņĪ░ĻĖ░ņŚÉ ļśÉļŖö ļ╣äņĀĢņāüņĀüņ£╝ļĪ£ ĒŖĖļ”ĮņŗĀņ£╝ļĪ£ ĒÖ£ņä▒ĒÖöļÉśļ®┤ ĒÖ£ņä▒ĒÖöļÉ£ ĒŖĖļ”ĮņŗĀņŚÉ ļČĆņ░®ļÉśņ¢┤ ĒŖĖļ”ĮņŗĀņØś ņ×æņÜ®ņØä ņ¢ĄņĀ£ĒĢśļŖö ļ░®ņ¢┤ ņŚŁĒĢĀņØä ļŗ┤ļŗ╣ĒĢ£ļŗż[31,32]. SPINK1ņØĆ N34S, P55S ļō▒ņØś ļ│ĆņØ┤ņŚÉ ņØśĒĢ┤ņä£ ĻĘĖ ĻĖ░ļŖźņØä ņāüņŗżĒĢśņŚ¼ ĻĖ░ļŖź ņåīņŗżņä▒ ļ│ĆņØ┤(loss-of-function mutation)Ļ░Ć ņ£Āļ░£ļÉśļŖöļŹ░, ņØ┤Ļ▓āņØĆ ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝Ļ│╝ Ļ╣ŖņØĆ ņŚ░Ļ┤ĆņØ┤ ņ׳ļŗż[23]. SPINK1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ļŖö ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝ļ┐Éļ¦ī ņĢäļŗłļØ╝ ņ£ĀņĀäņä▒ ņĘīņןņŚ╝Ļ│╝ ņĢīņĮöņś¼ņä▒ ņĘīņןņŚ╝ņŚÉņä£ļÅä Ļ┤Ćņ░░ļÉśļŖöļŹ░, ņØ┤ ļ│ĆņØ┤ ļŗ©ļÅģņ£╝ļĪ£ļŖö ņĘīņןņŚ╝ņØ┤ ņל ļ░£ņāØĒĢśņ¦Ć ņĢŖĻĖ░ ļĢīļ¼ĖņŚÉ ņĢ×ņŚÉņä£ ņ¢ĖĻĖēĒĢ£ ņĘīņןņŚ╝ņØś ņ¦üņĀæņĀüņØĖ ņøÉņØĖņ£╝ļĪ£ Ļ░äņŻ╝ĒĢśĻĖ░ ļ│┤ļŗżļŖö ņ£Āļ░£ņØĖņ×É(predisposing factor) ļśÉļŖö ĻĖ░ņŚ¼ņØĖņ×É(contributing factor)ļĪ£ ļ│┤ļŖö Ļ▓¼ĒĢ┤Ļ░Ć ņ¦Ćļ░░ņĀüņØ┤ļŗż[23].
ņ×ÉĻ░Ćļ®┤ņŚŁņä▒(autoimmune)
ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝(autoimmune pancreatitis)ņØĆ ņ×ÉĻ░Ćļ®┤ņŚŁĻĖ░ņĀäņŚÉ ņØśĒĢ£ ņĘīņןņØś ļ¦īņä▒ ņŚ╝ņ”Ø ņ¦łĒÖśņ£╝ļĪ£ ņĀäņ▓┤ ļ¦īņä▒ņĘīņןņŚ╝ņØś 2-4%ļź╝ ņ░©ņ¦ĆĒĢśļ®░, ņĪ░ņ¦üĒĢÖņĀü, ņśüņāüĒĢÖņĀü, ņŗżĒŚśņŗżĻ▓Ćņé¼ ļ░Å ņ×äņāüņåīĻ▓¼ņŚÉņä£ ļŗżļźĖ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ĻĄ¼ļ│äļÉśļŖö ĒŖ╣ņ¦ĢņĀüņØĖ ņ¢æņāüņØä ļ│┤ņØĖļŗż[33]. ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ type 1Ļ│╝ type 2ļĪ£ ļéśļē£ļŗż (Table 3) [34,35]. Type 1 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ņĘīņןļ┐É ņĢäļŗłļØ╝ ņĀäņŗĀņØä ņ╣©ļ▓öĒĢśņŚ¼ ļŗ┤Ļ┤Ć, Ēøäļ│Ąļ¦ē, ņ×äĒīīņäĀ, ņŗĀņן, ļłłļ¼╝ņāś, ņ╣©ņāś, Ļ░æņāüņäĀ, Ļ░ä ļō▒ņŚÉļÅä ņä¼ņ£ĀĒÖö ņŚ╝ņ”ØņØä ņ£Āļ░£ĒĢĀ ņłś ņ׳ņ£╝ļ®░, ņØ┤ņÖĆ Ļ░ÖņØĆ ņĀäņŗĀņןĻĖ░ ņ╣©ļ▓öņŚÉ ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░G4Ļ░Ć ņżæņÜöĒĢ£ ņŚŁĒĢĀņØä ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ļö░ļØ╝ņä£ type 1 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░G4ņŚ░Ļ┤Ćņä▒ ņĀäņŗĀņ¦łĒÖś(immunoglobulin G4-associated systemic disease)ņØ┤ ņĘīņןņŚÉņä£ ļ░£ĒśäļÉ£ Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│Ā ņ׳ļŗż. Type 2 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ļīĆļČĆļČäņØś Ļ▓ĮņÜ░ ņĘīņןņŚÉļ¦ī ņŚ╝ņ”ØņØ┤ ĻĄŁĒĢ£ļÉśĻ│Ā ļŗżļźĖ ņןĻĖ░ņØś ņ╣©ļ▓öņØ┤ ļō£ļ¼╝ņ¦Ćļ¦ī ņŚ╝ņ”Øņä▒ ņןņ¦łĒÖśĻ│╝ņØś ņŚ░Ļ┤Ćņä▒ņØĆ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż.
Type 1 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ļ”╝ĒöäĒśĢņ¦łņäĖĒż ņ╣©ņ£ż Ļ▓ĮĒÖöņä▒ ņĘīņןņŚ╝(lymphoplasmacytic sclerosing pancreatitis, LPSP)ņØ┤ļØ╝Ļ│ĀļÅä ļČĆļź┤ļ®░, ņĪ░ņ¦üĒĢÖņĀüņ£╝ļĪ£ ņĘīĻ┤ĆņŻ╝ņ£ä ļ”╝ĒöäĻĄ¼ ļ░Å ĒśĢņ¦łņäĖĒżņØś ņ╣©ņ£ż(periductal lymphoplasmacytic infiltrate), ņåīņÜ®ļÅīņØ┤ ļ¬©ņ¢æ Ēś╣ņØĆ ļéśņäĀĒśĢņØś ņä¼ņ£ĀĒÖö(swirling or storiform fibrosis), ņØ┤ņÖĆ ļÅÖļ░śļÉ£ ĒÅÉņćäņä▒ ņåīņĀĢļ¦źņŚ╝(obliterative venulitis), ĻĘĖļ”¼Ļ│Ā ņĘīņןļé┤ ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░ G4 ņ¢æņä▒ ĒśĢņ¦łņäĖĒżņØś ņ╣©ņ£ż(abundant IgG4 immunostainig) (> 10 cells/HPF)ņØä ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢ£ļŗż. ļśÉĒĢ£ ĒŖ╣ņ¦ĢņĀüņ£╝ļĪ£ Ēśłņ▓Ł ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░G4ņØś ņāüņŖ╣ņØä ļÅÖļ░śĒĢ£ļŗż. Type 2 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ļ╣äņĢīņĮöņś¼ņä▒ ņĘīĻ┤Ć ĒīīĻ┤┤ņä▒ ņĘīņןņŚ╝(non-alcoholic ductdestructive pancreatitis) ļśÉļŖö Ļ│╝ļ”ĮĻĄ¼ ņāüĒö╝ ļ│æļ│Ć(granulocyte epithelial lesion, GEL) ņ¢æņä▒ ņĘīņןņŚ╝ņ£╝ļĪ£ļÅä ļČłļ”¼ļ®░, ņĪ░ņ¦üĒĢÖņĀüņ£╝ļĪ£ ņżæņä▒ĻĄ¼ņØś ņĘīĻ┤Ćļ▓Į ņ╣©ņ£żĻ│╝ ĻĘĖļĪ£ ņØĖĒĢ£ ņĘīĻ┤Ćļ▓Į ņāüĒö╝ņØś ĒīīĻ┤┤ļź╝ ĒŖ╣ņ¦Ģņ£╝ļĪ£ ĒĢ£ļŗż. ņØ┤ Ļ▓ĮņÜ░ Ēśłņ▓Ł ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░G4ļŖö ņĀĢņāüņØ┤Ļ│Ā ļ®┤ņŚŁĻĖĆļĪ£ļČłļ”░G4ņØś ņĘīņן ņ╣©ņ£żļÅä Ļ▒░ņØś ņŚåļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż.
ņĢäņ¦ü ņĀä ņäĖĻ│äņĀüņ£╝ļĪ£ Ļ│ĄĒåĄļÉ£ ņ¦äļŗ©ĻĖ░ņżĆņØĆ ņŚåļŖö ņāüĒā£ņØ┤ļéś ņ¦ĆĻĖłĻ╣īņ¦Ć ļéśņś© ļ¬©ļōĀ ņ¦äļŗ©ĻĖ░ņżĆļōżņØĆ type 1 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØä ņ¦äļŗ©ĒĢśļŖöļŹ░ ņ┤łņĀÉņØ┤ ļ¦×ņČöņ¢┤ņĀĖ ņ׳Ļ│Ā, type 2 ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØä ņ¦äļŗ©ĒĢśĻĖ░ ņ£äĒĢ┤ņä£ Ēśäņ×¼ļĪ£ļŖö ņĪ░ņ¦üĒĢÖņĀü Ļ▓Ćņé¼Ļ░Ć ĒĢäņłś ņé¼ĒĢŁņØ┤ļŗż. ņ×ÉĻ░Ćļ®┤ņŚŁņä▒ ņĘīņןņŚ╝ņØĆ ņ×äņāüņĀüņ£╝ļĪ£ ņĘīņןņĢöĻ│╝ ĻĄ¼ļČäĒĢśĻĖ░ ĒלļōżĻ│Ā ņ£Āņé¼ĒĢ£ ņ”ØņāüņØä ļ│┤ņØ┤ļŖö Ļ▓ĮņÜ░Ļ░Ć ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņŻ╝ņØśļź╝ ņÜöĒĢ£ļŗż. ņ╣śļŻīļŖö ņŖżĒģīļĪ£ņØ┤ļō£ņØś Ļ▓ĮĻĄ¼ ļ│ĄņÜ®ņØä ĻĘ╝Ļ░äņ£╝ļĪ£ ņØ┤ļŻ©ņ¢┤ņ¦Ćļ®░, ņĢ×ņŚÉņä£ ņ¢ĖĻĖēĒ¢łļō»ņØ┤ ņäĖĒżņ¦ł ļé┤ļĪ£ ļ╣äņĀĢņāüņĀüņ£╝ļĪ£ ļČäĒżļÉśņŚłļŹś CFTRņØ┤ ņŖżĒģīļĪ£ņØ┤ļō£ņŚÉ ņØśĒĢ┤ ņĀĢņāüņĀüņ£╝ļĪ£ ņĘīņäĀļ░®ņäĖĒżņŚÉņä£ Ļ┤ĆĻ░Ģ ļé┤ļĪ£ Ē¢źĒĢśļŖö Ēæ£ļ®┤ņŚÉ ņ×¼ļČäĒżļÉśļŖö Ļ▓āņØ┤ ņŻ╝ņÜö ĻĖ░ņĀäņ£╝ļĪ£ ņāØĻ░üļÉśĻ│Ā ņ׳ļŗż[30].
ņ×¼ļ░£ņä▒ ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝(recurrent and severe acute pancreatitis associated chronic pancreatitis)
ĻĖēņä▒ņĘīņןņŚ╝ņØĆ ĒĢŁņāü ņĀĢņāüņ£╝ļĪ£ ĒÜīļ│ĄļÉśļŖö Ļ▓āņØĆ ņĢäļŗłļ®░, ņĢäņ¦üĻ╣īņ¦ĆļŖö ļģ╝ļ×ĆņØś ņŚ¼ņ¦ĆĻ░Ć ņ׳ņ£╝ļéś ņ×¼ļ░£ņä▒ ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ┤ĆĻ│äļŖö ņŚ¼ļ¤¼ ņŚ░ĻĄ¼ņÖĆ ņŗżĒŚś, ĒŖ╣Ē׳ ņ£ĀņĀäņä▒ ņĘīņןņŚ╝ņŚÉ Ļ┤ĆĒĢ£ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ņä£ ļŹöņÜ▒ ĒÖĢņŗżĒĢ┤ņ¦ĆĻ│Ā ņ׳ļŗż[6]. Whitcomb ļō▒ņØĆ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ļ│æĻĖ░ņĀäņ£╝ļĪ£ PRSS1 ņ£ĀņĀäņ×É ļ░Å SPINK1 ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņŚÉ ņØśĒĢ£ ņĘīņäĀļ░®ņäĖĒżļé┤ ĒŖĖļ”ĮņŗĀņØś ņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöņŚÉ ļö░ļźĖ ļ░śļ│ĄņĀüņØĖ ĻĖēņä▒ņĘīņןņŚ╝ ļśÉļŖö ņĢīņĮöņś¼ņŚÉ ņØśĒĢ£ ļ░śļ│ĄņĀüņØĖ ņĘīņäĖĒżņØś ņåÉņāüņØ┤ ņ┤łļלļÉśĻ│Ā ņØ┤ņŚÉ ņłśļ░śļÉ£ ņŚ╝ņ”ØņäĖĒżņØś ņ╣©ņ£żĻ│╝ ņŚ╝ņ”ØņäĖĒżņŚÉņä£ņØś ņŚ╝ņ”Øņä▒ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņØä ņāØņä▒ņØ┤ ņ£ĀļÅäļÉśĻ│Ā ņØ┤ņŚÉ ļö░ļźĖ ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöĻ░Ć ņØ┤ļŻ©ņ¢┤ņ¦Ćļ®┤ Ļ▓░Ļ│╝ņĀüņ£╝ļĪ£ ņĘīņןļé┤ ņä¼ņ£ĀĒÖöĻ░Ć ņ¦äĒ¢ēļÉśņ¢┤ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ ĻĘĆņ░®ļÉ£ļŗżļŖö ņØ┤ļźĖļ░ö SAPE (sentinel acute pancreatitis event) Ļ░ĆņäżņØä ņĀ£ņŗ£ĒĢśņśĆļŗż[25]. ņØ┤ņÖĆĻ░ÖņØ┤ ņĢīņĮöņś¼Ļ│╝ ņ£ĀņĀäņä▒ ņĘīņןņŚ╝ ņØ┤ņÖĖņŚÉļÅä ņŚ¼ļ¤¼ ņĢĮņĀ£, ĒØĪņŚ░, Ļ│Āņ¦ĆĒśłņ”Ø, Ļ│Āņ╣╝ņŖśĒśłņ”Ø, ļŗ┤ņäØ ļō▒ ĻĖēņä▒ņĘīņןņŚ╝ņØä ņ£Āļ░£ņŗ£Ēé¼ ņłś ņ׳ļŖö ļ¬©ļōĀ ņøÉņØĖņØĆ ņ×¼ļ░£ņä▒ ĻĖēņä▒ņĘīņןņŚ╝ņØś ņøÉņØĖņØ┤ ļÉĀ ņłś ņ׳ņ£╝ļ®░, Ļ▓░ĻĄŁņŚÉļŖö ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ ņ¦äĒ¢ēļÉĀ ņłś ņ׳ļŗż.
ĒÅÉņćäņä▒ ļ¦īņä▒ņĘīņןņŚ╝(obstructive chronic pancreatitis)
ĒÅÉņćäņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ĒÅÉņćä ļśÉļŖö Ēśæņ░® ĻĘ╝ņ£äļČĆņØś ņĘīĻ┤Ć ĒÖĢņן, ņĘīņäĀļ░®ņäĖĒż ņ£äņČĢ, ĻĘĖļ”¼Ļ│Ā ņĘīņŗżņ¦łņØś ļ»Ėļ¦īņä▒ ņä¼ņ£ĀĒÖöļØ╝ļŖö ĒśĢĒā£ĒĢÖņĀü ĒŖ╣ņ¦ĢņØä Ļ░¢ļŖöļŗż[36]. ņĘīĻ┤ĆņØś ĒÅÉņćä ļśÉļŖö Ēśæņ░®ņØś ņøÉņØĖņ£╝ļĪ£ļŖö Ļ░Ćņä▒ļéŁņóģĻ│╝ Ļ░ÖņØĆ ĻĖēņä▒ņĘīņןņŚ╝ņØś ĒĢ®ļ│æņ”Ø, ņÖĖņāü, ņóģņ¢æ, ņśżļööņö© Ļ┤äņĢĮĻĘ╝ ņØ┤ņāü, ļČäĒĢĀ ņĘīņן ļō▒ņØ┤ ņ׳ļŗż[6]. ņśżļööņö© Ļ┤äņĢĮĻĘ╝ ņØ┤ņāüņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØä ņØ╝ņ£╝ĒéżļŖöĻ░ĆņŚÉ ļīĆĒĢ┤ņä£ļŖö ļŹö ļ¦ÄņØĆ ņŚ░ĻĄ¼Ļ░Ć ĒĢäņÜöĒĢśņ¦Ćļ¦ī, ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ņśżļööņö© Ļ┤äņĢĮĻĘ╝ ņØ┤ņāüņØ┤ ņŚåļŖö ņé¼ļ×īņŚÉ ļ╣äĒĢ┤ ņ׳ļŖö ņé¼ļ×īņŚÉĻ▓īņä£ ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ ļÅÖļ░śļÉĀ ĒÖĢļźĀņØ┤ 4.6ļ░░ ņ”ØĻ░ĆĒĢ£ļŗżĻ│Ā ļ│┤Ļ│ĀĒĢśņśĆļŗż[37]. ļČäĒĢĀ ņĘīņןĻ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ┤ĆĻ│äļŖö ņĢäņ¦ü ļģ╝ļ×ĆņØś ņŚ¼ņ¦ĆĻ░Ć ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ņØś ņ¦äĒ¢ēņśłļ░®ņØä ņ£äĒĢ£ ļČĆņ£ĀļæÉ Ļ┤äņĢĮĻĘ╝ ņĀłĻ░£ņłĀ(minor papilla sphincterotomy)ņØś ņŗ£Ē¢ēņŚ¼ļČĆļŖö ņŗĀņżæņØä ņÜöĒĢ£ļŗż. ņĘīĻ┤Ć ĒÅÉņćäļź╝ ņĪ░ĻĖ░ņŚÉ ĻĄÉņĀĢĒĢ┤ ņżä Ļ▓ĮņÜ░ ņĪ░ņ¦üĒĢÖņĀü ĻĘĖļ”¼Ļ│Ā ĻĖ░ļŖźņĀü ļ│ĆĒÖöļź╝ ņ¢┤ļŖÉ ņĀĢļÅä ĒÜīļ│Ąņŗ£Ēé¼ ņłś ņ׳ņ£╝ļéś ņĘīņןņåÉņāüņØ┤ ņśüĻĄ¼ņĀüņ£╝ļĪ£ ļé©ĻĖ░ļÅäĒĢ£ļŗż.
ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ļ│æĻĖ░ņĀä(pathogenesis of chronic pancreatitis)
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņä¼ņ£ĀĒÖö ĻĖ░ņĀä
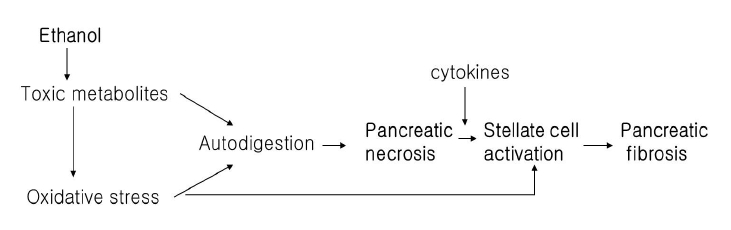
ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æļ”¼ĒĢÖņĀü ņåīĻ▓¼ņŚÉņä£ ņä¼ņ£ĀĒÖöļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØä ĻĖēņä▒ņĘīņןņŚ╝Ļ│╝ ĻĄ¼ļČä ņ¦ōļŖö ņżæņÜöĒĢ£ ļ│æļ”¼ ņåīĻ▓¼ņØ┤ļŗż. ņĘīņןņØś ņä¼ņ£ĀĒÖöļŖö Ļ░äņØś ņä¼ņ£ĀĒÖöņŚÉ Ļ┤ĆņŚ¼ĒĢśļŖö ņä▒ņāüņäĖĒż(stellate cell)ņÖĆ Ļ░ÖņØĆ ņ£ĀĒśĢņØś ņäĖĒżĻ░Ć ņĘīņן ļé┤ņŚÉņä£ ĻĘ£ļ¬ģļÉśĻ│Ā ņäĖĒżņØś ņäĖĒżņāØļ¼╝ĒĢÖņĀü ņä▒ņāüņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ē ļÉśļ®┤ņä£ ļ¦ÄņØĆ ļČĆļČäņØ┤ ĻĘ£ļ¬ģļÉśĻ│Ā ņ׳ļŗż. ņĘīņä▒ņāüņäĖĒżņØś ņĪ┤ņ×¼ļŖö ņØ╝ņ░ŹņØ┤ 1982ļģä Watari ļō▒ņØ┤ ņāØņźÉņŚÉņä£ ĻĘĖ ņĪ┤ņ×¼ļź╝ ļ│┤Ļ│ĀĒĢśņśĆļŗż[38]. 1990ļģäņŚÉļŖö Ikejiri ļō▒ņØ┤ ņĀäņ×ÉĒśäļ»ĖĻ▓Į ļ░Å ĒÄĖĻ┤æĒśäļ»ĖĻ▓ĮņØä ĒåĄĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ ņĘīņä▒ņāüņäĖĒżĻ░Ć ĒśłĻ┤Ć ņŻ╝ņ£ä ļ░Å ņäĀļ░®ņäĖĒż ņé¼ņØ┤ņŚÉ ņĪ┤ņ×¼ĒĢśļ®░ ņäĖĒżņ¦łļé┤ ĒśäņĀĆĒĢ£ ņ¦Ćļ░®Ļ│╝ļ”Į(lipid droplet)ņØä ĒĢ©ņ£ĀĒĢśļŖö Ļ▓ā ļō▒ņØś ņäĖĒżĒĢÖņĀü ĒŖ╣ņä▒ņØä Ļ░¢ļŖöļŗżĻ│Ā ĻĘ£ļ¬ģĒĢśņśĆļŗż[39]. ņĄ£ĻĘ╝ ApteņÖĆ Bachem ļō▒ņŚÉ ņØśĒĢ┤ ļ░▒ņä£ņÖĆ ņé¼ļ×īņŚÉņä£ ņä▒ņāüņäĖĒżĻ░Ć ļČäļ”¼ ļÅÖņĀĢ, ļ░░ņ¢æļÉ©ņŚÉ ļö░ļØ╝ ņäĖĒżņØś ņĪ░ņ¦ü ĒÖöĒĢÖņĀü ĒŖ╣ņä▒Ļ│╝ ņäĖĒżņāØļ¼╝ĒĢÖņĀü ĻĖ░ļŖźņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ĒÖ£ļ░£ĒĢ┤ņ¦ĆĻ│Ā ņ׳ļŗż[40,41]. ņĘīņä▒ņāüņäĖĒżļŖö ļ╣äĒÖ£ļÅÖĻĖ░ņŚÉļŖö ņäĖĒżņ¦łļé┤ ņżæņŗ¼ņŚÉ ĒĢĄņØä Ļ░¢Ļ│Ā ņ׳Ļ│Ā UVĻ┤æņŚÉ ļģ╣ņāēņØś ĒśĢĻ┤æņ£╝ļĪ£ ļ│┤ņØ┤ļŖö ļ╣äĒāĆļ»╝ AĻ░Ć ļŗżļ¤ē ĒĢ©ņ£ĀļÉ£ Ļ│ĄĒżļź╝ ņäĖĒżņ¦łņŚÉ Ļ░¢Ļ│Ā ņ׳ļŖö ņäĖĒżļĪ£ ņĘīņן ļé┤ņŚÉ ņĪ┤ņ×¼ĒĢ£ļŗż. ņØ┤ ņäĖĒżļź╝ ļČäļ”¼, ļÅÖņĀĢĒĢśņŚ¼ ņØ╝ņĀĢĻĖ░Ļ░ä ļ░░ņ¢æņØä ĒĢśļ®┤ ņäĖĒżņ¦łļé┤ ļ╣äĒāĆļ»╝A Ļ│╝ļ”ĮņØĆ ņåīņŗżļÉśĻ│Ā ╬▒-smooth muscle actin (╬▒-SMA)ņŚÉ ņŚ╝ņāēļÉśļŖö ņäĖĒżļĪ£ Ēæ£ĒśäĒśĢņØ┤ ļ│ĆĒÖöĒĢśĻ▓ī ļÉśņ¢┤ ļ¦łņ╣ś myofibroblastņ¢æ ņäĖĒżļĪ£ ņĀäĒÖśļÉśĻ▓ī ļÉ£ļŗż[40,41]. ņĘīņä▒ņāüņäĖĒżņØś ļ®┤ņŚŁĒÖöĒĢÖņĀü ņŚ░ĻĄ¼ņŚÉņä£ ņä▒ņāüņäĖĒżļŖö collagen type IĻ│╝ type IV, fibronectin, lamininĻ│╝ Ļ░ÖņØĆ ņäĖĒżĻ░äņ¦łļ¼╝ņ¦ł(extracelluar matrix, ECM)ņØä ĒĢ®ņä▒ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņĀĖ ņä¼ņ£ĀĒÖöņŚÉ ņżæņÜöĒĢ£ ĻĖ░ļŖźņØä ĒĢśļŖö ņäĖĒżļĪ£ ļ░øņĢäļōżņŚ¼ņ¦ĆĻ│Ā ņ׳ļŗż[41,42]. ņĘīņןļ┐É ņĢäļŗłļØ╝ ņŚ¼ļ¤¼ ņĪ░ņ¦üņØś ņä¼ņ£ĀĒÖö Ļ│╝ņĀĢņØĆ ECMņØś ĒĢ®ņä▒Ļ│╝ ļČäĒĢ┤ņØś ļČĆņĪ░ĒÖö ņ”ē ĒĢ®ņä▒ņØś ņ”ØĻ░ĆņÖĆ ļČäĒĢ┤ņØś ņĀĆĒĢśņŚÉ ņØśĒĢśņŚ¼ ņä¼ņ£ĀņØś ņ╣©ņ░®ņŚÉ ņØśĒĢ┤ ļ░£ņāØļÉ£ļŗż. ļö░ļØ╝ņä£ ņĘīņן ņä¼ņ£ĀĒÖöņŚÉ ņ׳ņ¢┤ņä£ ņĘīņä▒ņāüņäĖĒżņŚÉ ņØśĒĢ£ ECMņØś ņāØņä▒ļ┐Éļ¦ī ņĢäļŗłļØ╝ ECMņØś ļČäĒĢ┤ņŚÉ ņ׳ņ¢┤ ņĘīņä▒ņāüņäĖĒżņØś ņŚŁĒĢĀņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ļ¦ÄņØ┤ ņ¦äĒ¢ēļÉśĻ│Ā ņ׳ļŗż. ECMņØś ļČäĒĢ┤ņŚÉļŖö ņØ╝ļĀ©ņØś matrix metalloproteinase (MMPs)Ļ░Ć Ļ┤ĆņŚ¼ĒĢ£ļŗż. MMPsņØś ĒÖ£ņä▒ņØĆ TIMPs (tissue inhibitor of metaloproteinase)ņŚÉ ņØśĒĢśņŚ¼ ĻĖĖĒĢŁ ļÉśļŖöļŹ░ ņĢĮ 4Ļ░£ņØś ņĢäĒśĢņØ┤ ņĪ┤ņ×¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ņĘīņäĀļ░®ņäĖĒżņŚÉņä£ņØś MMP2ņØś ņĪ┤ņ×¼ ļ░Å TIMP1, TIMP2ņØś ņĪ┤ņ×¼Ļ░Ć ļ░ØĒśĆņĀĖ Ļ░äņä▒ņāüņäĖĒżņÖĆ Ļ░ÖņØ┤ ņĘīņןņØś ECMņØś ļČäĒĢ┤ļÅä ņä▒ņāüņäĖĒżĻ░Ć Ļ┤ĆņŚ¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņ¦ĆĻ│Ā ņ׳ļŗż[43]. ļśÉĒĢ£ ņĘīņä▒ņāüņäĖĒżļŖö ņä¼ņ£ĀņøÉņäĖĒżņÖĆ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ņØ┤ļÅÖ ļŖźļĀźņØ┤ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ĻĘ£ļ¬ģļÉśĻ│Ā ņ׳ļŗż[44]. ņŗżņĀ£ ņé¼ļ×īņØś ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ TNBS (trinitrobezene sulfonic acid) ļ░Å dibutyltin chloride ļō▒ņØś ņĢĮļ¼╝ņŚÉ ņØśĒĢ£ ļ¦īņä▒ņĘīņןņŚ╝ ļ¬©ļŹĖ, ņ×Éļ░£ņä▒ ņĘīņןņŚ╝ ļÅÖļ¼╝ļ¬©ļŹĖ(WBN/Kob rat), TGF╬▓1ņŚÉņä£ ņĘīņןņØś ņä¼ņ£ĀĒÖöņŚÉ ņä▒ņāüņäĖĒżņØś ņŚŁĒĢĀņØ┤ ņ”Øļ¬ģļÉśĻ│Ā ņ׳ļŗż[45-47]. ņ”ē ņä¼ņ£ĀĒÖöĻ░Ć ņØ╝ņ¢┤ļé£ Ļ││ņŚÉņä£ ĒÖ£ņä▒ ņä▒ņāüņäĖĒżĻ░Ć Ļ┤Ćņ░░ļÉśļ®░ in situ hybridisationĻĖ░ļ▓Ģņ£╝ļĪ£ ╬▒-SMAņ¢æņä▒ ņäĖĒżņŚÉņä£ procollagen mRNAĻ░Ć Ļ▓ĆņČ£ļÉ©ņ£╝ļĪ£ņŹ© collagen ņāØņä▒ņØ┤ ņä▒ņāüņäĖĒżņŚÉņä£ ņØ┤ļŻ©ņ¢┤ņ¦ĆļŖö Ļ▓āņ£╝ļĪ£ ņ”Øļ¬ģļÉśĻ│Ā ņ׳ļŗż[45].
ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ ņä¼ņ£ĀĒÖöļŖö ņ¢┤ļ¢╗Ļ▓ī ņ£Āļ░£ļÉśļŖöĻ░Ć?
ņĘīņןņŚÉņä£ņØś ņä¼ņ£ĀĒÖöļŖö ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ļÅģņä▒ ļ¼╝ņ¦łņŚÉ ņØśĒĢ┤ņä£ ļśÉļŖö ņĘīņן ņåÉņāüņŚÉ ņØśĒĢ┤ ņĘīņןņ£╝ļĪ£ļČĆĒä░ ņāØņä▒ļÉśļŖö ļ¼╝ņ¦łņŚÉ ņØśĒĢśņŚ¼ ņĘīņä▒ņāüņäĖĒżĻ░Ć ĒÖ£ņä▒ĒÖö ļÉśļ®░ ĒÖ£ņä▒ĒÖöļÉ£ ņä▒ņāüņäĖĒżņŚÉņä£ ECMņØś ņāØņä▒ņØ┤ ņ”ØĻ░Ć ļÉ©ņ£╝ļĪ£ņŹ© ņä¼ņ£ĀĒÖöĻ░Ć ļ░£ņāØĒĢ£ļŗż. ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ░Ćņן ĒØöĒĢ£ ņ£Āļ░£ņÜöņØĖņØĆ ņĢīņĮöņś¼ņØ┤ļŗż. Apte ļō▒ņØĆ ļ░░ņ¢æļÉ£ ņä▒ņāüņäĖĒżņŚÉ ņØ╝ņĀĢ ļåŹļÅäņØś ņŚÉĒāäņś¼ ļśÉļŖö ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ļź╝ Ēł¼ņŚ¼ĒĢśļ®┤ ņä▒ņāüņäĖĒżņŚÉņä£ ╬▒-SMAņØś ļ░£ĒśäņØ┤ ņ”ØĻ░Ć ĒĢśļ®░ ņĮ£ļØ╝Ļ▓ÉņØś ņāØņä▒ņØ┤ ņ”ØĻ░Ć ļÉ©ņØä Ļ┤Ćņ░░ĒĢśņśĆņ£╝ļ®░ ļ░░ņ¦ĆņŚÉ ņŚÉĒāäņś¼ņØś ņé░ĒÖöĻ│╝ņĀĢņØä ņ¢ĄņĀ£ĒĢśļŖö 4-methylpyrazoleņØä Ēł¼ņŚ¼ ĒĢśļ®┤ ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ņØś ņāØņä▒ ņ¢ĄņĀ£ņÖĆ ļŹöļČłņ¢┤ ņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöĻ░Ć ņ¢ĄņĀ£ ļÉ©ņØä ļ│┤Ļ│ĀĒĢśĻ│Ā ņ׳ļŗż[48]. ļśÉĒĢ£ ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ļŖö malondiadehydeņÖĆ Ļ░ÖņØĆ ņ£Āļ”¼ņé░ĒÖöĻĖ░ļź╝ ņ”ØĻ░Ćņŗ£ņ╝£ ņäĖĒż ļÅģņä▒ņØä ļéśĒāĆļé┤ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░ ĒĢŁņé░ĒÖöņĀ£ņØś ņØ╝ņóģņØĖ ļ╣äĒāĆļ»╝ EņØś Ēł¼ņŚ¼ ņŗ£ ņŚÉĒāäņś¼ ļ░Å ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ņŚÉ ņØśĒĢ£ ņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöĻ░Ć ņ¢ĄņĀ£ ļÉ©ņØä ļ│┤Ļ│ĀĒĢśĻ│Ā ņ׳ļŗż[48]. ļö░ļØ╝ņä£ ņŚÉĒāäņś¼ ļśÉļŖö ņŚÉĒāäņś¼ ļīĆņé¼ ļ¼╝ņ¦łņØĖ ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ņŚÉ ņØśĒĢśņŚ¼ ņĘīņä▒ņāüņäĖĒżĻ░Ć ņ¦üņĀæņĀüņ£╝ļĪ£ ĒÖ£ņä▒ĒÖöļÉśņ¢┤ ņĮ£ļØ╝Ļ▓É ļō▒ņØś ņä¼ņ£ĀĒÖöļź╝ ļ░£ņāØņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņŻ╝ņןļÉśĻ│Āņ׳ļŗż. ļśÉĒĢ£ ņĄ£ĻĘ╝ ņŚÉĒāäņś¼ ļ░Å ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ļŖö ņĘīņä▒ņāüņäĖĒżņŚÉņä£ ERK1/2, JNK, p38 kinase ļō▒ņØś MAP kinaseņØś ĒÖ£ņä▒ĒÖöņØś ņäĖĒżļé┤ ņŗĀĒśĖņĀäļŗ¼ ļ¼╝ņ¦łņØ┤ ņä▒ņāüņäĖĒżņŚÉņä£ņØś collagenņØś ĒĢ®ņä▒ņŚÉ Ļ┤ĆņŚ¼ ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņ¦ĆĻ│Ā ņ׳ļŗż[49].
ņĢīņĮöņś¼Ļ│╝ ņĘīņןĻĖ░ļŖźņØś ļ│ĆĒÖö
Ēśäņ×¼Ļ╣īņ¦Ć ņĢīņĮöņś¼ņØ┤ ņĘīņןņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņŚÉ ļīĆĒĢ£ ļ¦ÄņØĆ ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ēļÉśņ¢┤ ņÖöņ£╝ļ®░ Ļ░äļץĒ׳ Ēśäņ×¼Ļ╣īņ¦Ć ļ│┤Ļ│ĀļÉ£ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝ļź╝ ĻĖ░ņłĀĒĢśĻ│Āņ×É ĒĢ£ļŗż.
ņĘīĻ┤Ć ļé┤ņĢĢņØś ņāüņŖ╣
ņŚÉĒāäņś¼ņØĆ ņĘīĻ┤Ć ļé┤ņĢĢņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ņĘīĻ┤Ć ļé┤ņĢĢņØś ņāüņŖ╣ņØĆ ņĘīņן Ļ░äņ¦łļé┤ļĪ£ņØś ņĘīņĢĪņØś ņŚŁļźśļź╝ ņ┤łļלĒĢśņŚ¼ ņĘīņןņåīĒÖöĒÜ©ņåīņŚÉ ņØśĒĢ£ ņĘīņןņØś ņ×ÉĻ░ĆņåīĒÖöļź╝ ņ£Āļ░£ĒĢśĻ│Ā ĻĖ░ĒāĆļÅģņä▒ļ¼╝ņ¦łņØś ņĘīņן ļé┤ļĪ£ņØś ņĀæĻĘ╝ņØä ņÜ®ņØ┤ĒĢśĻ▓ī ĒĢśņŚ¼ ņĘīņןņåÉņāüņØä ņ┤łļלĒĢĀ ņłś ņ׳ĻĖ░ ļĢīļ¼ĖņŚÉ ņĘīņןņŚ╝ ļ░£ņāØņŚÉ ļ¦żņÜ░ ņżæņÜöĒĢ£ ņØĖņ×ÉņØ┤ļŗż. ņĘīĻ┤ĆņØś ļé┤ņĢĢņŚÉ ņśüĒ¢źņØä ļ»Ėņ╣śļŖö ņÜöņØĖņ£╝ļĪ£ļŖö ņĘīņĢĪņØś ļČäļ╣äļ¤ē ļ░Å ņĘīņĢĪņØś ņĀÉļÅä ļ░Å ņśżļööņö© Ļ┤äņĢĮĻĘ╝ņØś ņĢĢļĀź ļ│ĆĒÖö ļō▒ņØ┤ ņ׳ļŗż. ņĢīņĮöņś¼ņØ┤ ņśżļööņö© Ļ┤äņĢĮĻĘ╝ņØś ĻĖ░ņĀĆņĢĢņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ņ£╝ļéś ņśżļööņö© Ļ┤äņĢĮĻĘ╝ņØś ņĢĢļĀź ļ│ĆĒÖöņÖĆ ņĘīņןņŚ╝ņØś ļ░£ņāØĻ│╝ņØś ņŚ░Ļ┤Ćņä▒ņØĆ ņĢäņ¦ü ĒÖĢņŗżĒ׳ ĻĘ£ļ¬ģļÉśĻ│Ā ņ׳ņ¦Ć ļ¬╗ĒĢśļŗż[50,51]. ņĘīĻ┤Ć ļé┤Ēö╝ ņäĖĒżĻ░äņØś ņäĖĒżĻ▓░ĒĢ®ņØĆ ņäĀļ░®ņäĖĒżņŚÉņä£ ļČäļ╣äļÉ£ ņĘīņåīĒÖö ĒÜ©ņåīņØś ņĘīĻ░äņ¦łļé┤ļĪ£ņØś ņØ┤ļÅÖņØä ļ░®ņ¦ĆĒĢśļŖö ĻĖ░ļŖźņØä Ļ░¢Ļ│Ā ņ׳ļŗż. ņĄ£ĻĘ╝ņØś ņŚ░ĻĄ¼ņŚÉ ņØśĒĢśļ®┤ ņŚÉĒāäņś¼ņØĆ ņĘīĻ┤Ćļé┤Ēö╝ņäĖĒżĻ░äņØś ņäĖĒż Ļ▓░ĒĢ®ņØä ļŖÉņŖ©ĒĢśĻ▓ī ĒĢśņŚ¼ ņĘīĻ┤Ć ļé┤ņØś ņĘīņĢĪņØ┤ Ļ░äņ¦łļĪ£ņØś ņŚŁļźśļź╝ ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņ¦ĆĻ│Ā ņ׳ļŗż[52]. ņĘīņĢĪņØś ņĘīĻ░äņ¦ł ļé┤ļĪ£ņØś ņ╣©Ēł¼ļŖö Ļ│¦ ņĘīņןņØś ņåīĒÖöņåÉņāüņØä ņ£Āļ░£ĒĢśņŚ¼ ņĘīņןņŚ╝ņØś ļ░£ņāØņØä ņ┤łļלĒĢśĻ▓ī ļÉ£ļŗż. ĻĘĖļ¤¼ļéś ņĘīĻ┤ĆņØś Ēł¼Ļ│╝ļĀź ņ”ØĻ░Ć ļ¦īņ£╝ļĪ£ļŖö ņĘīņןņŚ╝ņØä ņ£Āļ░£ĒĢśņ¦Ć ņĢŖļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ņ¢┤ ļŗżļźĖ ĻĖ░ņĀäņØ┤ ļ│ĄĒĢ®ņĀüņ£╝ļĪ£ ņ×æņÜ®ĒĢśņŚ¼ ņĘīņןņŚ╝ņØä ņ£Āļ░£ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉśĻ│Ā ņ׳ļŗż.
ņĢīņĮöņś¼ņØś ņäŁņĘ©ļŖö ņŗżĒŚśļÅÖļ¼╝ņŚÉņä£ ņĘīņןņÖĖļČäļ╣äņŚÉ ļīĆĒĢśņŚ¼ ĒĢŁņ¦ä ļśÉļŖö ņ¢ĄņĀ£ĒĢśļŖö ĻĖ░ļŖźņØä ļ│┤ņØ┤ļŖö ņāüļ░śļÉ£ ņŚ░ĻĄ¼ Ļ▓░Ļ│╝Ļ░Ć ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż. ņ”ē ņĀĢņāüņØĖņŚÉņä£ ņĢīņĮöņś¼ņØś ņäŁņĘ©ļŖö ņĘīņןļČäļ╣äļź╝ ņ¢ĄņĀ£ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņ¦ĆĻ│Ā ņ׳ņ£╝ļéś ļ¦īņä▒ ņĢīņĮöņś¼ ņżæļÅģņ×ÉņØś Ļ▓ĮņÜ░ ņĢīņĮöņś¼ņØĆ ņĘīņןļČäļ╣äļź╝ ĒĢŁņ¦ä ņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[53].
ņĘīņĢĪņØś ļŗ©ļ░▒ņ¦ł ļåŹļÅäļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ ņŚåļŖö ļ¦īņä▒ ņØīņŻ╝ņ×ÉņŚÉņä£ ņ”ØĻ░Ć ļÉśļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŖöļŹ░ ņØ┤ļŖö ņĘīņןņåīĒÖöĒÜ©ņåīņØś ļČäļ╣ä ņ”ØĻ░ĆņÖĆ ņĘīņןņØś ņżæĒāäņé░ ļČäļ╣äņĢĪņØś Ļ░ÉņåīņŚÉ ĻĖ░ņØĖĒĢ£ļŗż[54]. ņØ┤ļ¤¼ĒĢ£ ņĘīņĢĪļé┤ ļŗ©ļ░▒ņ¦łļåŹļÅäņØś ņ”ØĻ░ĆļŖö ņĘīņןņĢĪņØś ņĀÉņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżĻ│Ā ņØ┤Ļ▓āņØĆ ņĘīņĢĪļé┤ ļŗ©ļ░▒ņĀäņØś ĒśĢņä▒ņØä ņĪ░ņןĒĢśĻ│Ā ļ¦Éņ┤ł ņĘīĻ┤ĆņØś ĒÅÉņāēņØä ņ£Āļ░£ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļ®░ ņØ┤ļ¤¼ĒĢ£ ņĘīĻ┤ĆņØś ĒÅÉņćäļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØä ņ┤łļל ĒĢĀ ņłś ņ׳ļŖö ņżæņÜöĒĢ£ ĻĖ░ņĀäņØś ĒĢśļéśļĪ£ ņØĖņŗØļÉśĻ│Ā ņ׳ļŗż[55].
ĻĘĖļ¤¼ļéś ņĢīņĮöņś¼ņØ┤ ņśżļööņö© Ļ┤äņĢĮĻĘ╝ņØś ņĢĢļĀź ņ”ØĻ░Ć, ņĘīĻ┤ĆņäĖĒżņØś ņĢĮĒÖöļĪ£ ņØĖĒĢ£ ņĘīĻ┤Ć Ēł¼Ļ│╝ļĀźņØś ņ”ØĻ░Ć, ņĘīņĢĪņØś ļŗ©ļ░▒ņ¦ł ļåŹļÅäņØś ņ”ØĻ░ĆņŚÉ ļö░ļźĖ ņĀÉļÅäņØś ļ│ĆĒÖöņŚÉ ļö░ļØ╝ ņĘīĻ┤Ć ļé┤ņĢĢņØś ņāüņŖ╣ņØ┤ ņ¢┤ļ¢╗Ļ▓ī ļ¦īņä▒ņĘīņןņŚ╝ņØä ņ£Āļ░£ĒĢśĻ▓ī ļÉśļŖöĻ░ĆņŚÉ ļīĆĒĢ£ ĻĖ░ņĀäņØĆ ņĢäņ¦ü ļČłļČäļ¬ģĒĢ£ ņāüĒā£ņØ┤ļŗż.
ņĢīņĮöņś¼Ļ│╝ ņĘīņןņåīĒÖöĒÜ©ņåī ĒĢ®ņä▒ ļ│ĆĒÖö
ļÅÖļ¼╝ņŗżĒŚśņŚÉņä£ ņĢīņĮöņś¼ņØś ņäŁņĘ©ļŖö ņĘīņןņŚÉņä£ ĒŖĖļ”Įņŗ£ļģĖĻ▓ÉĻ│╝ ņ╣┤ņØ┤ļ¬©ĒŖĖļ”ĮņŗĀņØś ĒĢ®ņä▒ņØĆ ņ”ØĻ░Ćņŗ£ĒéżļŖöļŹ░ ļ░śĒĢ┤ ĒĢŁĒŖĖļ”ĮņŗĀ ņ¢ĄņĀ£ņ×¼ņØś ĒĢ®ņä▒ ļČäļ╣äļŖö Ļ░Éņåīņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[56-58]. ļśÉĒĢ£ ļØ╝ņØ┤ņĪ░ņóĆĒÜ©ņåīļĪ£ ĒŖĖļ”ĮņŗĀļģĖĻ▓ÉņØś ĒÖ£ņä▒ĒÖöņÖĆ ņŚ░Ļ┤ĆņØ┤ ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņ¦ä cathepsin BņØś ĒĢ®ņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż. ņØ╝ļ░śņĀüņ£╝ļĪ£ ņĘīņןņŚÉņä£ ĒĢ®ņä▒ ļÉśļŖö ĒĢŁĒŖĖļ”ĮņŗĀ ņ¢ĄņĀ£ļ¼╝ņ¦łņØĆ ņĘīņןņŚÉņä£ ņāØņä▒ļÉśļŖö ĒŖĖļ”ĮņŗĀņØś ņĢĮ 10% ņĀĢļÅäļź╝ ņ¢ĄņĀ£ĒĢĀ ņłś ņ׳ļŖö ņ¢æņØ┤ ĒĢ®ņä▒ļÉśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņ¦ĆĻ│Ā ņ׳ļŖöļŹ░ ņĢīņĮöņś¼ ņäŁņĘ©ņŚÉ ņØśĒĢśņŚ¼ ņāüļīĆņĀüņ£╝ļĪ£ ĒŖĖļ”ĮņŗĀņØś ĒĢ®ņä▒ ļČäļ╣äļ¤ēņØ┤ ņ”ØĻ░ĆļÉśļ®┤ņä£ ņĘīņןļé┤ ņåīĒÖöĒÜ©ņåīņØś ņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöņØś Ļ░ĆļŖźņä▒ņØ┤ ļåÆņĢä ņĘīņןņŚ╝ņØś ļ░£ņāØĻ░ĆļŖźņä▒ļÅä ļåÆļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż.
ņĢīņĮöņś¼Ļ│╝ ņĘīņן Ēśłļźśļ¤ēņØś ļ│ĆĒÖöņÖĆ ņé░ĒÖöļ¼╝ņ¦ł
ņĘīņןņØś ĒŚłĒśł-ņ×¼Ļ┤ĆļźśņåÉņāüĻ│╝ ņé░ĒÖöļ¼╝ņ¦łņØĆ ĻĖēņä▒ņĘīņןņŚ╝ņØś ņżæņÜöĒĢ£ ļ░£ņāØ ĻĖ░ņĀä ņżæ ĒĢśļéśņØ┤ļŗż. ņĢīņĮöņś¼ņØĆ ņĘīņןņØś ĒśłņĢĪ ņł£ĒÖśļ¤ēņØä Ļ░Éņåīņŗ£ĒéżĻ│Ā ņØ┤ņŚÉ ļö░ļźĖ ņĘīņןņØś ĒŚłĒśłņåÉņāüņØ┤ ņ┤łļלļÉśļ®░ ņØ┤Ļ▓āņØ┤ ņĘīņןļé┤ ĒŖĖļ”ĮņŗĀņØś ņ×ÉĻ░Ć ĒÖ£ņä▒ĒÖöņÖĆ ņŚ░Ļ┤Ć ņ׳ļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[59]. ņĢīņĮöņś¼ņØĆ ņĘīņןņØś ņ£Āļ”¼ ņé░ņåīĻĖ░ņØś ņāØņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļÅÖļ¼╝ ņŗżĒŚśņŚÉņä£ ņ”Øļ¬ģļÉśĻ│Ā ņ׳ļŖöļŹ░ ņØ┤ņÖĆ Ļ░ÖņØĆ ĒŚłĒśłĻ│╝ ņ£Āļ”¼ ņé░ņåīĻĖ░ņØś ņ”ØĻ░ĆĻ░Ć ņĢīņĮöņś¼ņŚÉ ņØśĒĢ£ ņĘī ņåÉņāüņØś ņżæņÜöĒĢ£ ĻĖ░ņĀäņ£╝ļĪ£ ņ×æņÜ®ĒĢĀ Ļ░ĆļŖźņä▒ņØ┤ ļ¦żņÜ░ ļåÆļŗż.
ņĢīņĮöņś¼ņØś ņĘīņäĀļ░®ņäĖĒżņŚÉ ļīĆĒĢ£ ņ¦üņĀæņĀü ņ×æņÜ®
ņŚÉĒāäņś¼ņØĆ ņäĖĒżļ¦ēņØä ņĢĮĒÖöņŗ£ņ╝£ ņäĖĒżļé┤ ņåīĻĖ░Ļ┤ĆņØä ļČłņĢłņĀĢĒĢśĻ▓ī ļ¦īļōĀļŗż. ņāØņ▓┤ļ¦ēņØś ļČłņĢłņĀĢĒÖöļĪ£ ņØĖĒĢśņŚ¼ ņĘīņäĀļ░®ņäĖĒżļé┤ ļØ╝ņØ┤ņĪ░ņóĆņ£╝ļĪ£ļČĆĒä░ ļØ╝ņØ┤ņĪ░ņóĆ ĒÜ©ņåīņØĖ cathepsin BņØś ņ£ĀņČ£ņØä ņ£Āļ░£ĒĢśĻ│Ā ņØ┤ļĪ£ ņØĖĒĢ£ ņĘīņןļé┤ ņåīĒÖöĒÜ©ņåīņØś ņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöĻ░Ć ņ£Āļ░£ļÉĀ ņłś ņ׳ļŗż. ņŚÉĒāäņś¼ņØś ņāØņ▓┤ļ¦ē ļČłņĢłņĀĢĒÖö ņ×æņÜ®ņØĆ ņŚÉĒāäņś¼ņØ┤ļéś ņŚÉĒāäņś¼ņØś ļīĆņé¼ ļ¼╝ņ¦łņØĖ ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ņØś ņ¦üņĀæ ņ×æņÜ®ņØ┤ ņĢäļŗłļØ╝ ņŚÉĒāäņś¼ņŚÉ ņØśĒĢ£ ņĮ£ļĀłņŖżĒģīļĪżņŚÉņŖżĒä░ņØś ņČĢņĀü ļśÉļŖö ĒżņŖżĒżļ”¼ĒīīņĀ£ A2ņØś ĒÖ£ņä▒ņŚÉ ļö░ļźĖ Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[60,61]. ņŚÉĒāäņś¼ņØĆ ĒĢĄņé░ ļ░Å ņ╣╝ņŖś ņŗĀĒśĖ ņĀäļŗ¼Ļ│äņØś ĒÖ£ņä▒ĒÖöļź╝ ņ£ĀļÅäĒĢśĻ│Ā ņØ╝ļ░śņĀüņ£╝ļĪ£ Ļ│ĀļåŹļÅä(> 500 mM)ņŚÉņä£ Ļ┤Ćņ░░ļÉśļŖö Ļ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśĻ│Ā ņ׳ļŗż[62]. ņ”ē Ļ│ĀļÅÖļÅäņØś ņŚÉĒāäņś¼ņØĆ adenylate cyclaseņØä ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻ│Ā ņäĖĒżļé┤ ņ╣╝ņŖśļåŹļÅäļź╝ ņ”ØĻ░Ćņŗ£ņ╝£ ĒżņŖżĒżļ”¼ĒīīņĀ£ C ļō▒ņØä ĒÖ£ņä▒ĒÖöņŗ£ĒéżĻĖ░ ļĢīļ¼ĖņŚÉ ņäĖĒż ļé┤ņŚÉņä£ ĒÜ©ņåīņøÉ(zymogen)ņØś ĒÖ£ņä▒ĒÖöļŖö ņĘīņןņŚ╝ņØś ļ░£ļ│æĻĖ░ņĀäņ£╝ļĪ£ ļäÉļ”¼ ļ░øņĢäļōżņŚ¼ņ¦ĆĻ│Ā ņ׳ņ¦Ćļ¦ī ĒÜ©ņåīņøÉņØś ņ×ÉĻ░Ć ĒÖ£ņä▒ĒÖöņŚÉ ļīĆĒĢ£ ņĀĢĒÖĢĒĢ£ ņäĖĒżļé┤ ĻĖ░ņĀäņØĆ ņĢäņ¦ü ĻĘ£ļ¬ģļÉśņ¦Ć ņĢŖĻ│Ā ņ׳ļŗż. ĻĖēņä▒ņĘīņןņŚ╝ņØś ņŗżĒŚśļÅÖļ¼╝ļ¬©ļŹĖņŚÉņä£ CCKņØś Ļ│╝ļÅäĒĢ£ ņ×ÉĻĘ╣ņŚÉ ņØśĒĢ┤ ĒÜ©ņåīņøÉņØ┤ ņäĖĒżļé┤ ņ×ÉĻ░Ć ĒÖ£ņä▒ĒÖöĻ░Ć ņØ┤ļŻ©ņ¢┤ņ¦ĆļŖöļŹ░ ņŚÉĒāäņś¼ņØĆ ņØ┤ļ¤¼ĒĢ£ CCKņŚÉ ļīĆĒĢ£ Ļ░Éņłśņä▒ņØä ņ”ØĻ░Ćņŗ£ņ╝£ ņāØļ”¼ņĀüņØĖ ļåŹļÅäņØś CCKņŚÉ ņØśĒĢ┤ņä£ļÅä ĒÜ©ņåīņøÉņØś ĒÖ£ņä▒ĒÖöĻ░Ć ņØ┤ļŻ©ņ¢┤ņ¦ĆĻ▓ī ĒĢ£ļŗż[63]. ļśÉĒĢ£ ņŚÉĒāäņś¼ņØĆ ņŗŁņØ┤ņ¦ĆņןņŚÉņä£ CCKņØś ļČäļ╣äļź╝ ņ┤ēņ¦äņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ¢┤ ļæÉ Ļ░Ćņ¦Ć ĻĖ░ņĀäņŚÉ ņØśĒĢśņŚ¼ ņĘīņäĀļ░®ņäĖĒżļé┤ ņĘīņןĒÜ©ņåīņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöļź╝ ņĪ░ņןņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż. ņĢīņĮöņś¼ņØś ņĘīņן ļ░Å ņĘīņןņäĀļ░®ņäĖĒżņŚÉ ļīĆĒĢ£ ņäĖĒż ņāØļ¼╝ĒĢÖņĀü ņ×æņÜ®ņØĆ ņŗżĒŚś ļÅÖļ¼╝Ļ│╝ ņé¼ņÜ®ļåŹļÅä ļō▒ņŚÉ ļö░ļØ╝ ļŗżņ¢æĒĢśĻ▓ī Ļ┤Ćņ░░ļÉśļ»ĆļĪ£ ņĢäņ¦ü ļÜ£ļĀĘĒĢśĻ▓ī ņĀĢļ”ĮļÉśņ¦ĆļŖö ļ¬╗ĒĢśĻ│Ā ņ׳ļŗż.
ņĢīņĮöņś¼Ļ│╝ ļ¦īņä▒ņĘīņןņŚ╝
ņĢ×ņä£ ĻĖ░ņłĀĒĢ£ ļ░öņÖĆ Ļ░ÖņØ┤ ņŚÉĒāäņś¼ņØĆ ņĘīņןņŚÉņä£ ņĘīņן ņåīĒÖöĒÜ©ņåīņØś ĒĢ®ņä▒Ļ│╝ ļØ╝ņØ┤ņĪ░ņóĆņØś cathepsin BņØś ĒĢ®ņä▒ņØä ņ”ØĻ░Ćņŗ£ĒéżļŖö ļ░śļ®┤ ĒŖĖļ”ĮņŗĀ ņ¢ĄņĀ£ļ¼╝ņ¦łņØś ņāüļīĆņĀü ĒĢ®ņä▒ņØä Ļ░Éņåīņŗ£ĒéżĻ│Ā ņäĖĒżļ¦ē ļ░Å ņäĖĒżļé┤ ņåīĻĖ░Ļ┤ĆņØś ņāØņ▓┤ļ¦ēņØś ļČłņĢłņĀĢņŗ£ņ╝£ ņäĖĒżļé┤ ņåīĒÖöĒÜ©ņåīņØś ņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöļź╝ ņĪ░ņן ņŗ£ĒéżļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņ¦ĆĻ│Ā ņ׳ļŗż. ļö░ļØ╝ņä£ ļ░śļ│ĄņĀüņØĖ ņØīņŻ╝ņŚÉ ņØśĒĢ£ ļ░śļ│ĄņĀüņØĖ ņĘīņäĖĒżņØś ņåÉņāüņØ┤, ņØ┤ļźĖļ░ö Ļ┤┤ņé¼-ņä¼ņ£ĀĒÖöņäżņŚÉņä£ ņŻ╝ņןĒĢśļŖö Ļ▓āĻ│╝ Ļ░ÖņØ┤, ņĘīņäĀļ░®ņäĖĒżņØś ņåÉņāü ļ░Å ĻĘĖņŚÉ ļö░ļźĖ ņŚ╝ņ”ØņäĖĒżņØś ņ╣©ņ░®ņŚÉ ņØśĒĢśņŚ¼ ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöļź╝ ņ┤łļלĒĢĀ ņłś ņ׳ļŗż. ņŗżņĀ£ļĪ£ ņŗżĒŚśņĀü ņŚ░ĻĄ¼ņŚÉ ņØśĒĢśļ®┤, ņĘīņןņŚ╝ ļ¬©ļŹĖņŚÉņä£ ņĘīņןļé┤ ņ╣©ņ£żļÉ£ ņŚ╝ņ”ØņäĖĒż ļ░Å ņäĀļ░®ņäĖĒżļĪ£ļČĆĒä░ ļŗżņ¢æĒĢ£ ņŚ╝ņ”Øņä▒ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņØś ņāØņä▒ņØ┤ ņ”ØĻ░ĆļÉ£ļŗżļŖö Ļ▓āņØ┤ ņ×ģņ”ØļÉśņŚłļŗż. ļśÉĒĢ£ ņŗżĒŚśņĀüņ£╝ļĪ£ ņĘīņäĀļ░®ņäĖĒżļź╝ TNF-╬▒, interleukin-1 ļśÉļŖö interleukin-6 ļō▒ņØś ņŚ╝ņ”Øņä▒ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņ£╝ļĪ£ ņ▓śļ”¼ĒĢśļ®┤ ņäĀļ░®ņäĖĒżņØś ĒÖ£ņä▒ĒÖöņÖĆ ņä¼ņ£ĀņåīņØś ņāØņä▒ņØ┤ ņ”ØĻ░ĆļÉśļŖö Ļ▓āņ£╝ļĪ£ ļ░ØĒśĆņ¦ĆĻ│Ā ņ׳ļŗż. ĒŖ╣Ē׳ TGF╬▓1Ļ│╝ PDGF (platelet derived growth factor) ļō▒ņØĆ ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöņÖĆ ņ”ØņŗØ ņä¼ņ£ĀņåīņØś ņāØņä▒ņØä ņ£Āļ░£ņŗ£ĒéżļŖö ņżæņÜöĒĢ£ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņ£╝ļĪ£ ĻĘ£ļ¬ģļÉśĻ│Ā ņ׳ļŗż. ņóģĒĢ®ĒĢśļ®┤ ņĢīņĮöņś¼ņŚÉ ņØśĒĢ£ ņĘīņןņØś ņä¼ņ£ĀĒÖöļŖö ņŚÉĒāäņś¼ ļ░Å ĻĘĖ ļīĆņé¼ ļ¼╝ņ¦łņØĖ ņĢäņäĖĒŖĖņĢīļŹ░ĒĢśņØ┤ļō£ņŚÉ ņØśĒĢ£ ņ¦üņĀæņĀüņØĖ ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖö ļ░Å ņŚÉĒāäņś¼ņŚÉ ņØśĒĢ£ ņĘīņןļé┤ ņåīĒÖöĒÜ©ņåīņØś ņ×ÉĻ░ĆĒÖ£ņä▒ĒÖöņŚÉ ļö░ļźĖ ņĘīņäĖĒż ņåÉņāü ļ░Å ņŚ╝ņ”ØņäĖĒżņØś ņ╣©ņ£żņŚÉ ļö░ļź┤ļŖö ņŚ╝ņ”Øņä▒ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņØś ņāØņä▒Ļ│╝ ņØ┤ļ¤░ ņé¼ņØ┤ĒåĀņ╣┤ņØĖņŚÉ ņØśĒĢ£ ņØ┤ņ░©ņĀüņØĖ ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöņØś ņØ┤ņżæņĀüņØĖ ĻĖ░ņĀäņŚÉ ņØśĒĢśņŚ¼ ņ¦äĒ¢ēļÉśļŖö Ļ▓āņ£╝ļĪ£ ņČöņĀĢļÉśĻ│Ā ņ׳ļŗż(Fig. 2). ĻĘĖļ¤¼ļéś ņØ┤ļōż Ļ░ĆņäżņØĆ ļīĆļČĆļČä ņŗżĒŚśĻ┤Ćļé┤ ļśÉļŖö ļÅÖļ¼╝ ņŗżĒŚśņŚÉ ĻĘ╝Ļ▒░ĒĢ©ņ£╝ļĪ£ ļŹöņÜ▒ ļŹö ļ¦ÄņØĆ ņŚ░ĻĄ¼Ļ░Ć ņ¦äĒ¢ēļÉśņ¢┤ņĢ╝ ĒĢĀ Ļ▓āņØ┤ļŗż.
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝(natural history of chronic pancreatitis)
ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ņøÉņØĖ, ņ¦łļ│æņØś ļŗ©Ļ│ä, ĻĄŁņåī ĒĢ®ļ│æņ”Ø ņ£Āļ¼┤ņŚÉ ļö░ļØ╝ ļŗżņ¢æĒĢ£ ņ×äņāüņ¢æņāüņØä ļ│┤ņØ┤ļ»ĆļĪ£ ņ×ÉņŚ░Ļ▓ĮĻ│╝ļź╝ ĒīīņĢģĒĢśĻĖ░Ļ░Ć ņēĮņ¦Ć ņĢŖĻ│Ā, ņØ┤ņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ ļśÉĒĢ£ ņāüļīĆņĀüņ£╝ļĪ£ ļČĆņĪ▒ĒĢśļ®░, ņä£ļĪ£ ņāüļ░śļÉ£ Ļ▓░Ļ│╝ļōżļĪ£ ļģ╝ņ¤üņØ┤ ņ׳ņ¢┤ ņÖöļŗż[64]. ļö░ļØ╝ņä£ ļ│Ė Ļ│ĀņŚÉņä£ļŖö ņĢīņĮöņś¼Ļ│╝ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ ļīĆĒĢ┤ Ēśäņ×¼Ļ╣īņ¦Ć ļ░£Ēæ£ļÉ£ ļīĆĻĘ£ļ¬©ņØś ņĀäĒ¢źņĀü ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ļōżņØä ņżæņŗ¼ņ£╝ļĪ£, ņøÉņØĖņŚÉ ļö░ļźĖ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝ņŚÉ ļīĆĒĢśņŚ¼ Ļ│Āņ░░ĒĢ┤ļ│┤Ļ│Āņ×É ĒĢ£ļŗż.
ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝
ņŻ╝ņÜö ņ”ØņāüĻ│╝ ņ×ÉņŚ░ Ļ▓ĮĻ│╝
ĒåĄņ”Ø
ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉĻ▓ī Ļ░Ćņן Ēü░ ņ×äņāüņĀü ļ¼ĖņĀ£ļź╝ ņØ╝ņ£╝ĒéżļŖö Ļ▓āņØ┤ ĒåĄņ”ØņØ┤ļ®░, ļīĆļČĆļČä ĒÖśņ×ÉļŖö ņØ╝ņāüņāØĒÖ£ņŚÉ ņ¦ĆņןņØä ņ┤łļלĒĢĀ ņĀĢļÅäņØś ņŗ¼ĒĢ£ ĒåĄņ”ØņØä Ļ▓ĮĒŚśĒĢ£ļŗż. ņØ┤ļ¤¼ĒĢ£ ĒåĄņ”ØņŚÉ ļīĆĒĢ┤ ņ£Āļ│æĻĖ░Ļ░ä, ņĘīņן ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäņØś ņ¦äĒ¢ē ņĀĢļÅä, ņĘīņןļé┤ ņäØĒÜīĒÖö Ēś╣ņØĆ ņĘīĻ┤Ć ļ│ĆĒśĢ ļō▒ņØś ĒśĢĒā£ ļ│ĆĒÖöņÖĆ ņŚ░Ļ┤ĆļÉ£ ļ¦ÄņØĆ ņ×äņāü ņŚ░ĻĄ¼ļōżņØ┤ ņ׳ļŗż. ĒĢśņ¦Ćļ¦ī ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ņØś ĒåĄņ”ØņØĆ ņĘīĻ┤Ć Ēś╣ņØĆ ņĘīņן ņŗżņ¦ł ņĢĢļĀź ņāüņŖ╣ ņÖĖņŚÉļÅä ņĘīņןņŚÉ ļČäĒżĒĢśļŖö ņŻ╝ņ£ä ņŗĀĻ▓ĮņØś ņŚ╝ņ”Ø, ņĘīņן ņÖĖņĀüņØĖ ņÜöņåī ļō▒ ļŗżņ¢æĒĢ£ ĒåĄņ”Ø ņ£Āļ░£ ĻĖ░ņĀäņØ┤ ņĢīļĀżņĀĖ ņ׳ņ¢┤, ĒåĄņ”ØņØś ņ×äņāüĻ▓ĮĻ│╝ļź╝ ļŗ©ņł£Ē׳ ņĀĢĒśĢĒÖöĒĢśņŚ¼ ņäżļ¬ģĒĢśļŖö Ļ▓āņØĆ ļČłĻ░ĆļŖźĒĢśļŗż[65].
ĒåĄņ”ØņØś ņ£ĀĒśĢ
Ammann ļō▒ņØĆ 207ļ¬ģņØś ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×Éļź╝ ĒÅēĻĘĀ 17ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░ĒĢ£ ņĀäĒ¢źņĀü ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ļź╝ ĒåĄĒĢ┤ ļæÉ Ļ░Ćņ¦ĆņØś ĒŖ╣ņ¦ĢņĀüņØĖ ĒåĄņ”Ø ņ¢æņāüņØ┤ ņ׳ņØīņØä ņĀ£ņĢłĒĢśņśĆļŗż[66]. AĒśĢ ĒåĄņ”ØņØĆ ņ×¼ļ░£ņä▒ ĻĖēņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ĒØöĒ׳ Ļ┤Ćņ░░ļÉśļŖö Ļ▓āņ£╝ļĪ£ ĒåĄņ”Ø ņ¦ĆņåŹņŗ£Ļ░äņØ┤ ļ╣äĻĄÉņĀü ņ¦¦Ļ│Ā, 10ņØ╝ņØä ļäśĻĖ░ņ¦Ć ņĢŖņ£╝ļ®░ ņłś Ļ░£ņøöņŚÉņä£ ņłś ļģäņŚÉ ņØ┤ļź┤ļŖö ĒåĄņ”Øņåīņŗż Ē£┤ņ¦ĆĻĖ░Ļ░Ć ņĪ┤ņ×¼ĒĢ£ļŗż. ņØ┤ļ¤¼ĒĢ£ Ļ░äĒŚÉņĀü ņ¢æņāüņØä ļ│┤ņØ┤ļŖö AĒśĢ ĒåĄņ”ØņØĆ ļ¦īņä▒ņĘīņןņŚ╝ ņ×Éņ▓┤ņŚÉ ņØśĒĢ┤ ļ░£ņāØĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üĒĢ£ļŗż. ļ░śļ®┤ņŚÉ BĒśĢ ĒåĄņ”ØņØĆ ļ¦żņØ╝ ņ¦ĆņåŹļÉśļŖö ĒåĄņ”ØņØ┤ ņśżļ×£ ĻĖ░Ļ░ä ļéśĒāĆļéśĻ▒░ļéś ņżæņ”ØņØś ņ×¼ļ░£ņä▒ ĒåĄņ”ØņØ┤ ņŚ░ņåŹĒĢ┤ņä£ ļéśĒāĆļéśļŖö Ļ▓āņØ┤ ĒŖ╣ņ¦ĢņØ┤ļŗż. BĒśĢ ĒåĄņ”ØņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉļŖö ņØ╝ņŻ╝ņØ╝ņŚÉ 2ņØ╝ ņØ┤ņāü, ņĀüņ¢┤ļÅä 2Ļ░£ņøö ļÅÖņĢł ņŗ¼ĒĢ£ ĒåĄņ”ØņØä ĒśĖņåīĒĢśļ®░, ņØ┤ļŖö Ļ░Ćņä▒ļéŁņóģ, ļŗ┤ņ”ÖņÜĖĒśł, ņĘīĻ┤Ć ņĢĢļĀźņØś ņāüņŖ╣Ļ│╝ Ļ░ÖņØĆ ņĘīņןņŚ╝ņØś ĻĄŁņåī ĒĢ®ļ│æņ”Øņ£╝ļĪ£ ņØĖĒĢ┤ ņĢ╝ĻĖ░ļÉśļŖö Ļ▓āņ£╝ļĪ£ ņāØĻ░üĒĢ£ļŗż. ņØ┤ļ¤¼ĒĢ£ ļæÉ Ļ░Ćņ¦Ć ĒåĄņ”ØņØś ņ¢æņāüņØ┤ ļŗżļźĖ ņŚ░ĻĄ¼ņ×ÉļōżņŚÉ ņØśĒĢ┤ ĒÖĢņØĖļÉśņ¦Ć ļ¬╗Ē¢łļŗżļŖö Ļ▓āņØ┤ ņØ┤ ņŚ░ĻĄ¼ņØś Ļ░Ćņן Ēü░ ņĢĮņĀÉņØ┤ņ¦Ćļ¦ī ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ĒåĄņ”ØņØś ņ£ĀĒśĢņŚÉ ļö░ļØ╝ ļŗżļźĖ ņ×äņāüĻ▓ĮĻ│╝Ļ░Ć ņ׳Ļ│Ā, ĻĘĖņŚÉ ļö░ļØ╝ ņ╣śļŻīļÅä ļŗżļźĖ ļ░®ļ▓ĢņØä ņäĀĒāØĒĢ┤ņĢ╝ ĒĢ£ļŗżļŖö ņŗżļ¦łļ”¼ļź╝ ņżĆļŗżļŖö ņĀÉņŚÉņä£ ņØśņØśļź╝ Ļ░Ćņ¦ł ļ¦ī ĒĢśļŗż[65]. ņ”ē, AĒśĢ ĒåĄņ”ØņØĆ ļ│┤ņĪ┤ņĀüņ£╝ļĪ£ ņ╣śļŻīĒĢśļ®░ Ļ▓ĮĻ│╝ļź╝ ņé┤ĒÄ┤ļ│╝ ņłś ņ׳ņ¦Ćļ¦ī BĒśĢ ĒåĄņ”ØņØĆ ĻĄŁņåī ĒĢ®ļ│æņ”ØĻ│╝ Ļ┤ĆļĀ©ļÉśļ»ĆļĪ£ ļ░░ņĢĪļ▓ĢĻ│╝ Ļ░ÖņØĆ ņłśņłĀņĀü ļ░®ļ▓Ģņ£╝ļĪ£ ņ╣śļŻīĒĢ┤ņĢ╝ ĒĢ£ļŗżļŖö Ļ▓āņØ┤ ņØ┤ļōżņØś ņŻ╝ņןņØ┤ņŚłļŗż[64].
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ£Āļ│æĻĖ░Ļ░äĻ│╝ ĒåĄņ”ØņØś Ļ┤ĆĻ│ä
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äĒ¢ēņŚÉ ļö░ļØ╝ ņĀÉņ¦äņĀüņØĖ ņĘīņן ņŗżņ¦łņØś ĒīīĻ┤┤ļĪ£ ĒåĄņ”ØņØ┤ ņåīņŗżļÉ£ļŗżļŖö ŌĆ£burn-outŌĆØĻ░ĆņäżņØĆ ņśżļל ņĀäļČĆĒä░ ļģ╝ļ×ĆņØ┤ ņ׳ņ¢┤ ņÖöļŗż[65]. ĒĢ£ ļ│┤Ļ│ĀņŚÉ ļö░ļź┤ļ®┤ ņ£Āļ│æĻĖ░Ļ░äņØ┤ ĻĖĖņ¢┤ņ¦ÉņŚÉ ļö░ļØ╝ ĒĢ®ļ│æņ”ØņØ┤ ņŚåļŖö ļ¦ÉĻĖ░ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ļŖö ņ×ÉņŚ░ņĀüņ£╝ļĪ£ ĒåĄņ”ØņØ┤ ņåīņŗżļÉśļ®░, ĻĄŁņåī ĒĢ®ļ│æņ”ØņØ┤ ļéśĒāĆļé¼ņØä ļĢī ņłśņłĀņĀü ļ░®ļ▓Ģņ£╝ļĪ£ ĒĢ┤Ļ▓░ĒĢ£ ĒÖśņ×ÉļōżņŚÉņä£ļÅä ļ│æņØś ņ¦äĒ¢ēņŚÉ ļö░ļØ╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ĒåĄņ”ØņØ┤ ņåīņŗżļÉśņ¢┤, Ļ▓░ĻĄŁ ĒåĄņ”Ø ņ¢æņāüņŚÉ ņżĆĒĢ┤ ņĀüņĀłĒĢ£ ņ╣śļŻī ņäĀĒāØņØä Ē¢łņØä ļĢī ļ¬©ļōĀ ĒÖśņ×ÉļōżņØ┤ 6ļģä ņØ┤ļé┤ņŚÉ 50%, 10ļģä ņØ┤ļé┤ņŚÉ 80% ņØ┤ņāüņŚÉņä£ ĒåĄņ”ØņØ┤ ĒĢ┤ņåīļÉśņŚłļŗż[66]. ļśÉĒĢ£ 249ļ¬ģņØś ĒÖśņ×Éļź╝ 14.6-18.1ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆņØä ļĢī ĒÅēĻĘĀ 15ļģä ņØ┤ļé┤ņŚÉ 77%ņŚÉņä£ ĒåĄņ”ØņØś Ļ░Éņåīļéś ņåīņŗżņØ┤ ņ׳ņŚłļŗż[67]. ĒĢśņ¦Ćļ¦ī ņØ┤ņÖĆ ļŗżļź┤Ļ▓ī Lankish ļō▒ņØĆ 335ļ¬ģņØś ĒÖśņ×Éļź╝ 10ļģä ņØ┤ņāü ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆņØä ļĢī 65%ņŚÉņä£ ņŚ¼ņĀäĒ׳ ļ░śļ│ĄņĀüņØĖ ĒåĄņ”ØņØ┤ ļéśĒāĆļé©ņØä ļ│┤Ļ│ĀĒĢśņśĆļŗż[68]. Ļ▓░ĻĄŁ ņŗ£Ļ░äņØś Ļ▓ĮĻ│╝ņŚÉ ļö░ļØ╝ ņØ╝ļČĆ ĒÖśņ×ÉĻĄ░ņŚÉņä£ ĒåĄņ”ØņØś ĒśĖņĀäņØ┤ļéś ņåīņŗżņØ┤ ļéśĒāĆļéĀ ņłś ņ׳ņ£╝ļéś, ņØ┤ļŖö Ļ░£Ļ░£ņØĖņŚÉ ņ׳ņ¢┤ ņśłņĖĪņØ┤ ļČłĻ░ĆļŖźĒĢśļ»ĆļĪ£ ļ»ĖļלņŚÉ ņ׳ņØä ĒåĄņ”ØņØś ĒśĖņĀäņØä ņśłņāüĒĢśĻ│Ā ļ│┤ņĪ┤ņĀüņØĖ ņ╣śļŻīļ¦īņØä ņ£Āņ¦ĆĒĢśļŖö Ļ▓āņØĆ ļ¦łņĢĮņä▒ ņ¦äĒåĄņĀ£ņŚÉ ļīĆĒĢ£ ņżæļÅģ Ļ░ĆļŖźņä▒ ļō▒ņØä ņāØĻ░üĒ¢łņØä ļĢī ļ░öļ×īņ¦üĒĢ£ ļ░®ļ▓ĢņØ┤ ņĢäļŗłļŗż[65].
ņĘīņן ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäņØś ņ¦äĒ¢ēĻ│╝ ĒåĄņ”ØņØś Ļ┤ĆĻ│ä
ņĘīņן ņÖĖļČäļ╣ä ļ░Å ļé┤ļČäļ╣ä ĻĖ░ļŖź Ļ░ÉņåīņŚÉ ļö░ļØ╝ ĒåĄņ”ØņØ┤ Ļ░ÉņåīĒĢśļŖöĻ░ĆņŚÉ ļīĆĒĢ£ Ļ▓āļÅä ņ£äņØś ļ¼ĖņĀ£ņÖĆ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ņāüļ░śļÉ£ ņŚ░ĻĄ¼Ļ▓░Ļ│╝ļōżņØ┤ ņ׳ļŗż. ņŖżņ£äņŖżņØś ņŚ░ĻĄ¼ļōżņØĆ ņ£Āļ│æĻĖ░Ļ░äņØś ņ”ØĻ░ĆņŚÉ ļö░ļźĖ ņĘīņןļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖźņØś Ļ░ÉņåīņÖĆ ņŚ░Ļ┤ĆļÉśļŖö ĒåĄņ”ØņØś Ļ░Éņåīļź╝ ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņŻ╝ņןĒĢśņśĆļŗż[66,69]. ĒĢśņ¦Ćļ¦ī ņä£ņ¢æņØś ļŗżļźĖ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ņŗ¼ĒĢ£ ņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäņØ┤ ņ׳ļŖö ĒÖśņ×Éļōż ņżæ 57%, ņØĖņŖÉļ”░ ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢ£ ņŗ¼ĒĢ£ ļŗ╣ļć©ļ│æņØä ņĢōĻ│Ā ņ׳ļŖö ĒÖśņ×Éļōż ņżæ 59%ņŚÉņä£ ĒåĄņ”ØņØ┤ ņ¦ĆņåŹļÉśĻ│Ā ņ׳ņØīņØä ņ¦ĆņĀüĒĢśņśĆļŗż[68]. Ļ▓░ĻĄŁ ņĘīņן ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäņØś ņ¦äĒ¢ēņØ┤ ĒåĄņ”ØņØś Ļ▓ĮĻ│╝ņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØĆ ņ£Āļ│æĻĖ░Ļ░äĻ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ņĀ£ĒĢ£ņĀüņØ┤ļØ╝Ļ│Ā ĒĢĀ ņłś ņ׳Ļ▓Āļŗż.
ņĘīņןņØś ĒśĢĒā£ļ│ĆĒÖö(ņĘīņןņØś ņäØĒÜīĒÖö, ņĘīĻ┤ĆņØś ļ│ĆĒśĢ)ņÖĆ ĒåĄņ”ØņØś Ļ┤ĆĻ│ä
ņĘīņןņØś ņäØĒÜīĒÖöļŖö ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝ ņżæ Ēøäļ░śņŚÉ ļéśĒāĆļéśļ®░, ņ¦äļŗ© Ēøä 5.5-8.7ļģäņŚÉ ļ░£ņāØĒĢ£ļŗżļŖö ņĀÉņŚÉņä£ ņ¦äĒ¢ēļÉ£ ļ¦īņä▒ņĘīņןņŚ╝ņØä ņŗ£ņé¼ĒĢ£ļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż[67,69]. ĒĢśņ¦Ćļ¦ī ņĘīņןņØś ņäØĒÜīĒÖöļŖö Ļ│ĀņĀĢņĀüņØĖ Ļ▓āņØ┤ ņĢäļŗłļØ╝ ļÅÖņĀüņØĖ Ļ▓ĮĻ│╝ļź╝ Ļ░Ćņ¦ĆļŖöļŹ░, ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äĒ¢ēņŚÉ ļö░ļźĖ ņäØĒÜīĒÖöņØś ņĪ┤ņ×¼ņÖĆ ĻĘĖņŚÉ ļö░ļźĖ ĒåĄņ”ØņØś ņåīņŗż(85%)ņØä ļ░£Ēæ£Ē¢łļŹś ĒĢ£ ņŚ░ĻĄ¼ļŖö ņØ┤Ēøä Ļ┤Ćņ░░ĻĖ░Ļ░äņØ┤ ĻĖĖņ¢┤ņ¦ÉņŚÉ ļö░ļØ╝ 1/3ņĀĢļÅäņŚÉņä£ ņäØĒÜīĒÖöĻ░Ć ņåīņŗżļÉśĻ▒░ļéś Ļ░ÉņåīĒĢśļŖöĻ▓āņØä ļŗżņŗ£ ļ│┤Ļ│ĀĒĢśņśĆļŗż[69,70]. ļŗżļźĖ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ĒåĄņ”ØņØś ņåīņŗżņØ┤ ņäØĒÜīĒÖöĻ░Ć ņĪ┤ņ×¼ĒĢśļŖö ņĘīņןņŚ╝ ĻĄ░ņŚÉņä£ ļŹöņÜ▒ ļåÆņĢśņ¦Ćļ¦ī, ņäØĒÜīĒÖöĻĄ░ļÅä 56%ņŚÉņä£ ņ×¼ļ░£ļÉśļŖö ĒåĄņ”ØņØ┤ ņŚ¼ņĀäĒ׳ ņĪ┤ņ×¼ĒĢśņśĆļŗż[68]. ļö░ļØ╝ņä£ ņäØĒÜīĒÖöņØś ņ£Āļ¼┤ļ¦īņØä Ļ░Ćņ¦ĆĻ│Ā ĒåĄņ”ØņØś ņĀüĻ│Ā ļ¦ÄņØīņØä ņŻ╝ņןĒĢĀ ņłśļŖö ņŚåļŗż.
ņŻ╝ņĘīĻ┤ĆņØä ĒżĒĢ©ĒĢ£ ņĘīĻ┤ĆņØś ļ│ĆĒśĢ ņĀĢļÅäņÖĆ ĒåĄņ”ØĻ░äņØś ņāüĻ┤ĆĻ┤ĆĻ│äļÅä ļÜ£ļĀĘĒĢśņ¦Ć ņĢŖļŗż. ņØ┤ļŖö ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ ĒåĄņ”Ø ļ░£ņāØņØ┤ ņĘīĻ┤ĆņØś ļ│ĆĒśĢņ£╝ļĪ£ ļīĆĒæ£ļÉśļŖö ņĘīĻ┤Ć ņĢĢļĀź ņāüņŖ╣ ļĢīļ¼ĖņØ┤ļØ╝ļŖö ĻĖ░ņĀä ņØ┤ņÖĖņŚÉļÅä ļŗżņ¢æĒĢ£ ņøÉņØĖņØä Ļ░Ćņ¦ĆĻ│Ā ņ׳ņØīņŚÉņä£ ņĢī ņłś ņ׳ļō»ņØ┤ ņŚ¼ļ¤¼ ņ×äņāüņŚ░ĻĄ¼ļōżņŚÉņä£ ņĘīĻ┤ĆņØś ļ│ĆĒśĢĻ│╝ ĒåĄņ”Ø ļ░£ņāØ ņé¼ņØ┤ņŚÉ ņØ╝ņĀĢĒĢ£ ņāüĻ┤ĆĻ┤ĆĻ│äļź╝ ļ│┤ņŚ¼ņŻ╝ņ¦Ć ļ¬╗Ē¢łļŗż[65]. 64ļ¬ģņØś ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ĒåĄņ”ØņØś ņĀĢļÅäņÖĆ ERCP, CTņāüņØś ņżæņ”ØļÅäļŖö ņØśļ»Ė ņ׳ļŖö ņāüĻ┤ĆĻ┤ĆĻ│äĻ░Ć ņŚåņŚłĻ│Ā, ERCPņāü ņ¦äĒ¢ēļÉ£ ņżæņ”ØņØś ņĘīĻ┤Ć ļ│ĆĒÖöļź╝ ļ│┤ņØ┤ļŖö ĒÖśņ×ÉņØś ļŗ©ņ¦Ć 62%ņŚÉņä£ļ¦ī ņŗ¼ĒĢ£ ĒåĄņ”ØņØä ĒśĖņåīĒĢśņśĆļŗż[71]. ļŗżļźĖ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ERCPņāü ņĀĢņāü ņĘīĻ┤ĆņØä ļ│┤ņØ┤ļŖö ĒÖśņ×ÉņØś 71%ņŚÉņä£ ņśżĒ׳ļĀż ņ¦ĆņåŹņĀüņØĖ ĒåĄņ”ØņØä ĒśĖņåīĒĢśĻ│Ā ņ׳ņ¢┤ ņŚŁņŗ£ ņØ╝ņĀĢĒĢ£ ņŚ░Ļ┤Ćņä▒ņØ┤ ņŚåņŚłļŗż[68].
Ļ▓░ĻĄŁ ņĘīņןņØś ņäØĒÜīĒÖöļéś ņĘīĻ┤ĆņØś ļ│ĆĒśĢ Ļ░ÖņØĆ ĒśĢĒā£ĒĢÖņĀü ļ│ĆĒÖöļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØś ņĀäļ░śņĀüņØĖ ņśłĒøäļéś ĒåĄņ”ØņØś Ļ▓ĮĻ│╝ļź╝ ņśłņĖĪĒĢśļŖö ļŹ░ ņ׳ņ¢┤ ņóŗņØĆ ņ¦ĆĒæ£Ļ░Ć ņĢäļŗłļŗż.
Ļ░£Ļ░£ņØĖ ĒÖśņ×ÉņŚÉ ņ׳ņ¢┤ ĒåĄņ”ØņØś Ļ▓ĮĻ│╝ņŚÉ ļīĆĒĢ£ ņĀäļ░śņĀüņØĖ ņśłņĖĪņØĆ ļČłĻ░ĆļŖźĒĢśļ®░ ņĘīņן ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖźņØś Ļ░Éņåīļéś ņĘīņןņØś ĒśĢĒā£ļ│ĆĒÖöļĪ£ ĒåĄņ”ØņØś Ļ▓ĮĻ│╝ļź╝ ņśłņĖĪĒĢśĻĖ░ļŖö Ēלļōżļŗż. ĒĢśņ¦Ćļ¦ī ņ£Āļ│æĻĖ░Ļ░äņØ┤ ĻĖĖņ¢┤ņ¦łņłśļĪØ ĒåĄņ”ØņØś ņ×ÉņŚ░ņĀü ņåīņŗżņØ┤ ņØ╝ņ¢┤ļéśļŖö ĻĄ░ņØ┤ ņ׳ļŗżļŖö Ļ▓āņØĆ ņé¼ņŗżņØ┤ļ®░, ļ│┤ļŗż ņ¦ĆņåŹņĀüņØĖ ĒåĄņ”ØņØś ĒśĖņåīļŖö ĻĄŁņåī ĒĢ®ļ│æņ”ØņØś ļ░£ņāØņØä ņŗ£ņé¼ĒĢśļ»ĆļĪ£ ļ│┤ļŗż ņĀüĻĘ╣ņĀüņØĖ ņ¦äļŗ©Ļ│╝ ņ╣śļŻīĻ░Ć ĒĢäņÜöĒĢĀ Ļ▓āņØ┤ļŗż.
ņĘīņן ņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀä
ņל ņĢīļĀżņ¦ä Ļ▓āņ▓śļ¤╝ ņĘīņןņØĆ ņāüļŗ╣ĒĢ£ ņÖĖļČäļ╣ä ĻĖ░ļŖźņØś ņśłļ╣äļĀźņØä Ļ░Ćņ¦ĆĻ│Ā ņ׳ņ¢┤ņä£ ņ×öļźś ņÖĖļČäļ╣äĻĖ░ļŖźņØ┤ 10% ļ»Ėļ¦īņ£╝ļĪ£ Ļ░ÉņåīĒĢśņ¦Ć ņĢŖļŖö ĒĢ£ ņ¦Ćļ░®ļ│ĆņØĆ ļ░£ņāØĒĢśņ¦Ć ņĢŖņ£╝ļ®░[72], ļŹöņÜ▒ņØ┤ ņÜ░ļ”¼ļéśļØ╝ņØś Ļ▓ĮņÜ░ ļé┤ļČäļ╣ä ĻĖ░ļŖź ņĀĆĒĢśļĪ£ ļŗ╣ļć©Ļ░Ć ņ┤łļלļÉśņŚłņØīņŚÉļÅä ņ¦Ćļ░®ļ│ĆņØä ĒśĖņåīĒĢśļŖö ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉļōżņØĆ Ļ▒░ņØś ņŚåļŗż[15]. Ļ░Ćļüö ņŗ¼ĒĢ£ ņ¦Ćļ░®ļ│Ć ļĢīļ¼ĖņŚÉ ņ▓┤ņżæĻ░Éņåīļéś Ļ░ÉņŚ╝ņØ┤ ļ¼ĖņĀ£Ļ░Ć ļÉśļŖö Ļ▓ĮņÜ░ļŖö ņ׳ņ¦Ćļ¦ī ņØ┤ļ¤¼ĒĢ£ ņÖĖļČäļ╣ä ĻĖ░ļŖź ņןņĢĀļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ▓ĮĻ│╝ļéś ņśłĒøäņŚÉ Ēü░ ņśüĒ¢źņØä ņŻ╝ņ¦ĆļŖö ļ¬╗ĒĢ£ļŗż[73].
ņĘīņןņØś ņÖĖļČäļ╣ä ĻĖ░ļŖźņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äĒ¢ē Ļ▓ĮĻ│╝ņŚÉ ļö░ļØ╝ ņĢģĒÖöļÉśļŖöņ¦Ć ļśÉĒĢ£ ļģ╝ļ×ĆņØ┤ ņ׳ļŗż. ņĀÉņ¦äņĀüņØĖ ņÖĖļČäļ╣ä ĻĖ░ļŖźņØś Ļ░ÉņåīĻ░Ć ĒÅēĻĘĀ 5.6ļģä ĒøäņŚÉ 87%ņŚÉņä£ ņØ╝ņ¢┤ļéśĻĖ░ļÅä ĒĢśņ¦Ćļ¦ī ĒÅēĻĘĀ 13.1ļģäņØś ļŹö ĻĖ┤ ņŗ£Ļ░äņØ┤ Ļ▒Ėļ”¼ļŖö Ļ▓ĮņÜ░ļÅä ņ׳ņŚłļŗż[67,69]. ĒĢśņ¦Ćļ¦ī ļÅģņØ╝ņØś ļ│┤Ļ│ĀņŚÉņä£ļŖö 3.5-6.5ļģäņØś ĻĖ░Ļ░ä ļÅÖņĢł ļīĆņāüĒÖśņ×ÉņØś 46%ņŚÉņä£ļŖö ņÖĖļČäļ╣ä ĻĖ░ļŖźņØś ņĀĆĒĢśĻ░Ć ņŚåņŚłĻ│Ā, 43%ņŚÉņä£ļ¦ī ĻĖ░ļŖźņØś ņĀĆĒĢśĻ░Ć ņ¦äĒ¢ēļÉśņŚłņ£╝ļ®░, 11%ņŚÉņä£ļŖö ņśżĒ׳ļĀż ĻĖ░ļŖź ĒśĖņĀäņØ┤ ļéśĒāĆļé¼ļŗż[68]. Ļ▓░ĻĄŁ ņÖĖļČäļ╣äņןņĢĀ ĒÅēĻ░ĆņØś ļŗżņ¢æĒĢ£ ļ░®ļ▓ĢļōżĻ│╝ Ļ░üĻĖ░ ļŗżļźĖ ņČöņĀüĻ┤Ćņ░░ ĻĖ░Ļ░äņŚÉ ļö░ļźĖ ļģ╝ļ×ĆņØ┤ ņ׳ņ£╝ļéś, ļ│æņØś ņ¦äĒ¢ēņŚÉ ļö░ļØ╝ ņÖĖļČäļ╣äĻĖ░ļŖźņØś ņןņĢĀļŖö 50-80% ņĀĢļÅäņŚÉņä£ ļ░£ņāØĒĢĀ Ļ▓āņ£╝ļĪ£ ņāØĻ░üĒĢ£ļŗż[72].
ņĘīņן ļé┤ļČäļ╣ä ĻĖ░ļŖź ļČĆņĀä
ļŗ╣ļć©ļ│æņØĆ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņŻ╝ņÜö ĒøäĻĖ░ ĒĢ®ļ│æņ”Øņ£╝ļĪ£ ņéČņØś ņ¦łņŚÉ ņśüĒ¢źņØä ņżä ļ┐Éļ¦ī ņĢäļŗłļØ╝ ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņé¼ļ¦ØņŚÉ ĻĖ░ņŚ¼ĒĢśļŖö ļÅģļ”ĮņĀüņØĖ ņ£äĒŚśņØĖņ×ÉļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[74]. ņĘīņן ņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäĻ│╝ļŖö ļŗ¼ļ”¼ ņżæņ”Ø ņĀĆĒśłļŗ╣ Ļ░ÖņØĆ ņāØļ¬ģņØä ņ£äĒśæĒĢśļŖö ļ│æļ░£ņ”ØņØä ņĢ╝ĻĖ░ĒĢśĻ│Ā, ļŗżļźĖ ļŗ╣ļć©ļ│æ ĒÖśņ×ÉņŚÉņä£ņÖĆ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ļ»ĖņäĖ ļ░Å ļīĆ ĒśłĻ┤ĆĒĢ®ļ│æņ”ØņØ┤ ļ░£ņāØĒĢśļ®░[75], ņ×ÉņŚ░Ē׳ ļ¦īņä▒ņĘīņןņŚ╝ ņ£Āļ│æĻĖ░Ļ░ä Ēś╣ņØĆ Ļ┤Ćņ░░ ĻĖ░Ļ░äņØ┤ ĻĖĖņłśļĪØ ļŗ╣ļć© ĒĢ®ļ│æņ”ØļÅä ļ¦ÄņĢäņ¦äļŗż[73].
500ļ¬ģņØś ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉļōżņØä ņĘīņןņłśņłĀĻĄ░Ļ│╝ ļ╣äņłśņłĀĻĄ░ņ£╝ļĪ£ ļéśļłäņ¢┤ ņĢĮ 7ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆņØä ļĢī ļŗ╣ļć©ļ│æņØś ļ░£ņāØņ£äĒŚśņØ┤ ņĘīņןņłśņłĀĻĄ░ņØ┤ ņłśņłĀĒĢśņ¦Ć ņĢŖņØĆ ĻĄ░ņŚÉ ļ╣äĒĢ┤ ņ”ØĻ░ĆĒĢśņ¦ĆļŖö ņĢŖņĢśņ£╝ļéś, ņĘīņן ņøÉņ£äļČĆ ņĀłņĀ£ņłĀņØä ļ░øņØĆ ĻĄ░ņØĆ ņĘīņŗŁņØ┤ņ¦Ćņן ņĀłņĀ£ņłĀ, ņĘīņן ļ░░ņĢĪņłĀ Ēś╣ņØĆ ļéŁņóģ ļ░░ņĢĪņłĀ ļō▒ņØś ņłśņłĀņØä ļ░øņØĆ ĒÖśņ×Éļ│┤ļŗż ļŗ╣ļć©ļ│æ ļ░£ļ│æņØ┤ ļåÆņĢśļŗż. ņĘīņן ļ░░ņĢĪ ļ¬®ņĀüņØś ņłśņłĀņØ┤ ļŗ╣ļć©ļ│æņØś ļ░£ņāØņØä ņśłļ░®ĒĢśņ¦ĆļŖö ļ¬╗Ē¢łĻ│Ā, ļŗ╣ļć©ņØś ļ░£ņāØņ£äĒŚśņØĆ ņĘīņןņŚ╝ņØ┤ ņ¦äĒ¢ēĒĢĀņłśļĪØ ņ”ØĻ░ĆĒĢśņŚ¼, ņĘīņן ņäØĒÜīĒÖöĻ░Ć ņŗ£ņ×æļÉśļ®┤ 3ļ░░ ņØ┤ņāüņØś ņ£äĒŚśļÅäļź╝ ļ│┤ņśĆļŗż[74].
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äĒ¢ē Ļ▓ĮĻ│╝ņŚÉ ļö░ļźĖ ļé┤ļČäļ╣ä ĻĖ░ļŖźņØś ņĢģĒÖöļŖö ļīĆņ▓┤ņĀüņ£╝ļĪ£ ļ¬©ļōĀ ņĀäĒ¢źņĀü ņŚ░ĻĄ¼ņŚÉņä£ ņØ╝ņ╣śĒĢśļŖö Ļ▓░Ļ│╝ļź╝ ļ│┤ņśĆļŗż. 335ļ¬ģņØś ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×Éļź╝ ņĢĮ 10ļģäĻ░ä ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆņØä ļĢī ņ┤łĻĖ░ņŚÉļŖö ņżæļō▒ļÅä Ēś╣ņØĆ ņżæņ”ØņØś ļŗ╣ļć©ļ│æņØ┤ 8%ņŚÉ ļČłĻ│╝ĒĢśņśĆņ£╝ļéś 10ļģä ĒøäņŚÉļŖö ņŚ░ĻĄ¼ ņ┤łĻĖ░ņØś ņĢĮ 10ļ░░ņŚÉ ņØ┤ļź┤ļŖö 78%ņØś ĒÖśņ×ÉņŚÉņä£ ņżæļō▒ļÅä Ēś╣ņØĆ ņżæņ”ØņØś ļŗ╣ļć©ļ│æņ£╝ļĪ£ ļ░£ņĀäĒĢśņśĆļŗż[68]. ļŗżļźĖ ņŚ░ĻĄ¼ļōżņŚÉņä£ļÅä ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ļŗ╣ļć©ļ│æņØś ļ░£ņāØĻ╣īņ¦Ć Ļ░üĻ░ü 6-10ļģä, 19.8ļģäņ£╝ļĪ£ ĻĖ░Ļ░äņØś ņ░©ņØ┤ļŖö ņ׳ņŚłņ¦Ćļ¦ī ņĘīņןņŚ╝ņØś ņ¦äĒ¢ēņŚÉ ļö░ļźĖ ļé┤ļČäļ╣ä ĻĖ░ļŖźņØś ņĢģĒÖöļŖö ļÅÖņØ╝ĒĢśĻ▓ī ļéśĒāĆļé¼ļŗż[66,67,69].
ņ×äņāü ļŗ©Ļ│ä(clinical stage)
Ļ│╝Ļ▒░ņŚÉļŖö ņĢīņĮöņś¼ņä▒ ĻĖēņä▒ņĘīņןņŚ╝ņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØä ĻĖ░ņĀĆņ¦łĒÖśņ£╝ļĪ£ Ļ░Ćņ¦ĆĻ│Ā ņ׳ļŖö ĒÖśņ×ÉņŚÉņä£ ļ░£ņāØĒĢ£ļŗżĻ│Ā ņāØĻ░üĒĢśņśĆļŗż. ņØ┤ļ¤¼ĒĢ£ Ļ░ĆņäżņØĆ ņ▓śņØī ņĘīņןņŚ╝ņØ┤ ļ░£ņāØĒĢ£ ĒÖśņ×ÉņŚÉņä£ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņĪ░ņ¦üĒĢÖņĀü, ļ░®ņé¼ņäĀĒĢÖņĀü ņ”ØĻ▒░Ļ░Ć ņĪ┤ņ×¼ĒĢ£ļŗżļŖö ņŚ░ĻĄ¼ļōżņØä ĻĘ╝Ļ▒░ļĪ£ ĒĢśņśĆļŗż[76]. ĒĢśņ¦Ćļ¦ī ņĄ£ĻĘ╝ņØś ņŚ¼ļ¤¼ ņ×äņāü ļ░Å ņŗżĒŚśņŚ░ĻĄ¼ļź╝ ļ░öĒāĢņ£╝ļĪ£ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äĒ¢ēņŚÉ ļīĆĒĢ┤ņä£ļŖö ŌĆ£Ļ┤┤ņé¼-ņä¼ņ£ĀĒÖö(necrosis-fibrosis)ŌĆØ Ļ░ĆņäżņØ┤ ļ│┤ļŗż ņŗĀļ╣Öņä▒ ņ׳Ļ▓ī ļ░øņĢäļōżņŚ¼ņ¦ĆĻ│Ā ņ׳ļŖöļŹ░, ņØ┤ļŖö ļ░śļ│ĄņĀüņØĖ ĻĖēņä▒ņĘīņןņŚ╝ņØś ļ░£ņāØņØ┤ ņĘīņןņĪ░ņ¦üņŚÉ Ļ│äņåŹņĀüņØĖ ņåÉņāüņØä ņŻ╝Ļ│Ā, Ļ▓░ĻĄŁ ļ╣äĻ░ĆņŚŁņĀüņØĖ ņä¼ņ£ĀĒÖöļź╝ ņ┤łļלĒĢ£ļŗżļŖö Ļ▓āņØ┤ļŗż[76,77]. ņĘīņןņØś ņä¼ņ£ĀĒÖöļŖö ņĘīņןĻĖ░ļŖźņØś ņĀÉņ¦äņĀüņØĖ Ļ░ÉņåīņÖĆ ņŚ░Ļ┤ĆņØ┤ ņ׳ņŚłĻ│Ā, ĻĖēņä▒ņŚÉņä£ ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ņØś ņØ┤Ē¢ēņØĆ ĻĖēņä▒ņĘīņןņŚ╝ņØś ļ╣łļÅä ļ░Å ņżæņ”ØļÅäņÖĆ ļ░ĆņĀæĒĢ£ Ļ┤ĆĻ│äĻ░Ć ņ׳ņŚłļŗż[78,79]. ļÅÖļ¼╝ ņŗżĒŚśņŚÉņä£ļŖö ļ░śļ│ĄņĀüņØĖ ĻĖēņä▒ ĻČżņé¼ņä▒ ņŚ╝ņ”ØņØä ņ£ĀļÅäĒĢśņśĆņØä ļĢī ņĘīņןņØś ņä¼ņ£ĀĒÖöĻ░Ć ļ░£ņāØĒĢ©ņØä ļ│┤ņŚ¼ņŻ╝ņŚłļŗż[80].
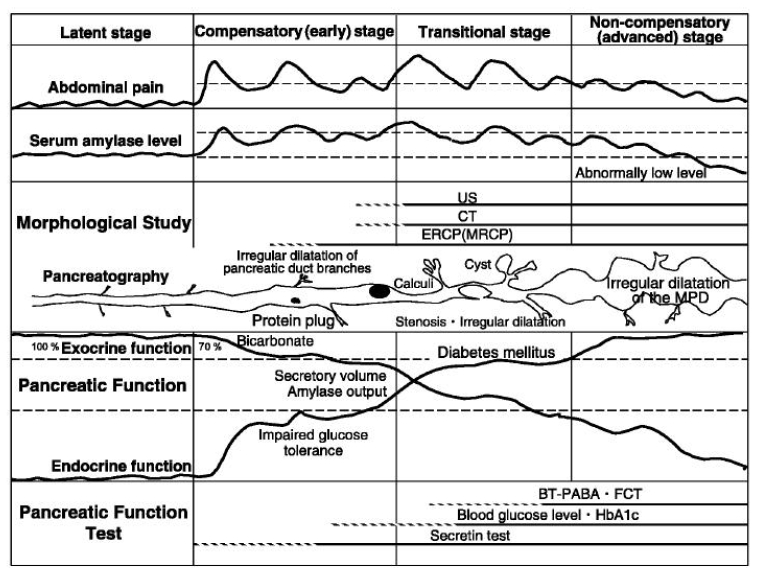
ņØ┤ļ¤¼ĒĢ£ Ļ┤┤ņé¼-ņä¼ņ£ĀĒÖö Ļ░ĆņäżĻ│╝ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ£Āļ│æĻĖ░Ļ░äņŚÉ ļö░ļźĖ ņĀÉņ¦äņĀüņØĖ ĻĖ░ļŖźņĀĆĒĢśņÖĆ ĒåĄņ”ØņåīņŗżņØä ļ│┤ņŚ¼ņŻ╝ņŚłļŹś ņŚ░ĻĄ¼ļōżņØä ĻĘ╝Ļ░äņ£╝ļĪ£ ĒĢśņŚ¼, ņ×äņāüņŚÉņä£ Ļ▓¬Ļ▓ī ļÉśļŖö ļŗżņ¢æĒĢ£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉļōżņØś Ļ┤Ćļ”¼ņŚÉ ņØ╝ņĀĢĒĢ£ ņ¦Ćņ╣©ņØä ņŻ╝ĻĖ░ ņ£äĒĢ┤ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×äņāüĻ▓ĮĻ│╝ļź╝ ļŗ©Ļ│äļ│äļĪ£ ņĀĢĒśĢĒÖöĒĢśņŚ¼ ļéśļłäņ¢┤ļ│┤ļĀżļŖö ņŗ£ļÅäļōżņØ┤ ņ׳ņ¢┤ņÖöļŗż(Fig. 3) [75,81-83]. ņĢ×ņä£ ņé┤ĒÄ┤ļ│┤ņĢśļō»ņØ┤ ņ£Āļ│æĻĖ░Ļ░äņŚÉ ļö░ļźĖ ĒåĄņ”ØņØś ņåīņŗż, ĒåĄņ”ØĻ│╝ ņĘīņןņØś ĻĖ░ļŖźņĀü, ĒśĢĒā£ĒĢÖņĀü ļ│ĆĒÖöņÖĆņØś ņŚ░Ļ┤Ć Ļ┤ĆĻ│ä, ĻĘĖļ”¼Ļ│Ā ņĘīņןĻĖ░ļŖźņØś ņĀÉņ¦äņĀüņØĖ ņĀĆĒĢś ļō▒ņŚÉ ļīĆĒĢ┤ņä£ ļģ╝ļ×ĆņØ┤ ņ׳Ļ│Ā ļ¬©ļōĀ ĒÖśņ×ÉļōżņØä ņØ┤ņÖĆ Ļ░ÖņØĆ ņ×äņāüļŗ©Ļ│äņŚÉ ņĀüņÜ®ņŗ£Ēé¼ ņłśļŖö ņŚåņ¦Ćļ¦ī, ņØ┤ļ¤¼ĒĢ£ ņ×äņāü ļŗ©Ļ│äņØś ĻĄ¼ļČäņØĆ ņŗżņĀ£ļĪ£ ĒÖśņ×ÉņØś ņ”ØņāüņØä Ļ│Āļ»╝ĒĢśĻ│Ā ņ╣śļŻīĒĢśļŖö ņ×äņāüņØśņŚÉĻ▓ī ņØ╝ņĀĢĒĢ£ ņŗżļ¦łļ”¼ļź╝ ņĀ£Ļ│ĄĒĢĀ ņłś ņ׳ņ£╝ļ®░, ĻĘĖ ņĀĢņØśņŚÉņä£ ļ╣äĻ░ĆņŚŁņä▒ņØä ļé┤ĒżĒĢśļŖö ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ░£ļģÉņŚÉ ņŚŁņäżņĀüņØ┤Ļ▓īļÅä Ļ░ĆņŚŁņĀüņØĖ ņ×äņāü ļŗ©Ļ│äĻ░Ć ņ׳ņØīņØä ņĀ£ņŗ£ĒĢśĻ│Ā ņ׳ļŗżļŖö ņĀÉņŚÉņä£ ņØśņØśĻ░Ć ņ׳ļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż.
ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝
ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æņØĖņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ņŚÉ ļö░ļØ╝ ņāłļĪ£ņÜ┤ ņ£ĀņĀäņĀü, ĒÖśĻ▓ĮņĀü, ļīĆņé¼ņä▒ ņøÉņØĖļōżņØ┤ ļ░ØĒśĆņ¦Ćļ®┤ņä£ ņØ┤ņĀäņŚÉ ĒŖ╣ļ░£ņä▒ņ£╝ļĪ£ ļČäļźśļÉśļŹś Ļ▓āļōżņØś ļ╣äņżæņØ┤ Ļ░ÉņåīĒĢśĻ│Ā ņ׳ņ¦Ćļ¦ī ņŚ¼ņĀäĒ׳ 10-30%ņŚÉņä£ļŖö ņøÉņØĖņØä ņĀĢĒÖĢĒ׳ ņĢī ņłś ņŚåļŗż[6,75]. ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ļ░£ļ│æļéśņØ┤ņŚÉ ļö░ļźĖ ĒĢśņ£ä ņ¦æļŗ©ņØ┤ ņĪ┤ņ×¼ĒĢśļ®░ ņØ┤ļōżņØĆ ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ļŖö ļŗżļźĖ ņ×ÉņŚ░Ļ▓ĮĻ│╝ļź╝ Ļ░Ćņ¦äļŗżļŖö Ļ▓āņØ┤ ņ▓śņØīņ£╝ļĪ£ ļ│┤Ļ│ĀļÉ£ ņØ┤Ēøä[84], Layer ļō▒ņØĆ ĒŖ╣ļ░£ņä▒ņŚÉ ļīĆĒĢ┤ ļ│┤ļŗż ņŚäĻ▓®ĒĢ£ ņ¦äļŗ©ĻĖ░ņżĆņØä ņĀüņÜ®ĒĢśņŚ¼ Ļ░üĻ░ü ļŗżļźĖ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØ┤ļŖö ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØś ĒĢśņ£ä ņ¦æļŗ©ņØä ĒÖĢņØĖĒĢśņśĆļŗż[67]. ņØ┤ļōżņØĆ ņ¢æļ®┤ļČäĒżņØś ĒśĖļ░£ ņŚ░ļĀ╣ļīĆļź╝ ļ│┤ņØ┤ļ®░ ļ░£ļ│æļéśņØ┤ņŚÉ ļö░ļØ╝ ļæÉ Ļ░Ćņ¦ĆļĪ£ ļéśļēśņ¢┤ņ¦äļŗż.
ņĪ░ĻĖ░ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝
ņØ┤ļōżņØś ļ░£ļ│æ ĒÅēĻĘĀļéśņØ┤ļŖö 19ņäĖļĪ£ 20ļīĆļź╝ ņĀäĒøäĒĢśņŚ¼ ļéśĒāĆļé£ļŗż. ļé©ņä▒ņŚÉņä£ ņŻ╝ļĪ£ ļéśĒāĆļéśļŖö ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ ļ╣äĒĢ┤ ļé©ļģĆ ņ░©Ļ░Ć ņŚåņ£╝ļ®░, ņ¦äļŗ© ņ▓śņØīļČĆĒä░ ļīĆļČĆļČäņØś ĒÖśņ×ÉņŚÉĻ▓ī ņŗ¼ĒĢ£ ĒåĄņ”ØņØ┤ ļéśĒāĆļéśļŖö Ļ▓āņØ┤ ņĢīņĮöņś¼ņä▒ ļ░Å ĒøäĻĖ░ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ĻĄ¼ļČäļÉśļŖö ĒŖ╣ņ¦ĢņØ┤ļŗż. ĒĢśņ¦Ćļ¦ī ņĘīņןņØś ņäØĒÜīĒÖö, ņĘīņן ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖźņØś ņĀĆĒĢśļŖö ņ¦äļŗ© ļŗ╣ņŗ£ņŚÉļŖö ļ¦żņÜ░ ļō£ļ¼╝Ļ│Ā(< 10%), ņ£Āļ│æĻĖ░Ļ░äņŚÉ ļö░ļźĖ ņĘīņןņŚ╝ņØś ĒśĢĒā£ĒĢÖņĀü, ĻĖ░ļŖźņĀü ļ│ĆĒÖöļÅä ļ¦żņÜ░ ļŖÉļ”¼Ļ▓ī ņ¦äĒ¢ēļÉ£ļŗż. ņĘīņןņØś ņäØĒÜīĒÖö ļ░£ņāØĻ╣īņ¦Ć ĒÅēĻĘĀ ņŗ£Ļ░äņØ┤ ņĢīņĮöņś¼ņä▒ ņĘīņןņŚ╝ņØś Ļ▓ĮņÜ░ļŖö 8.7ļģäņØĖļŹ░ ļ╣äĒĢ┤, ĒøäĻĖ░ ĒŖ╣ļ░£ņä▒, ņĪ░ĻĖ░ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ Ļ░üĻ░ü 19.4ļģä, 24.9ļģäņØä ļ│┤ņśĆļŗż. ļśÉĒĢ£, ņĘīņן ņÖĖļČäļ╣äĻĖ░ļŖź ļČĆņĀäĻ│╝ ļŗ╣ļć©ļ│æņØś ļ░£ņāØ ĻĖ░Ļ░äļÅä ņĢīņĮöņś¼ņä▒ ņĘīņןņŚ╝ņØś Ļ▓ĮņÜ░ļŖö Ļ░üĻ░ü 13.1ļģä, 19.8ļģäņØĖļŹ░ ļ╣äĒĢ┤, ĒøäĻĖ░ ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝ņØĆ 16.9ļģä, 11.9ļģäņ£╝ļĪ£, ņĪ░ĻĖ░ ĒŖ╣ļ░£ņä▒ ņĘīņןņŚ╝ņØĆ 26.3ļģä, 27.5ļģäņ£╝ļĪ£ ļ│┤Ļ│ĀļÉśņŚłļŗż[67].
ĒøäĻĖ░ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝
ņØ┤ļōżņØĆ ĒÅēĻĘĀ 56ņäĖņŚÉ ļ░£ļ│æĒĢśļ®░ ņĪ░ĻĖ░ĒśĢĻ│╝ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ļé©ļģĆĻ░äņØś ļ░£ņāØļźĀ ņ░©ņØ┤ļŖö ņŚåļŗż. ņĢīņĮöņś¼ņä▒, ņĪ░ĻĖ░ ĒŖ╣ļ░£ņä▒ņŚÉ ļ╣äĒĢ┤ ļ¦ÄņØĆ ņłśņŚÉņä£ ĒåĄņ”ØņØ┤ ņŚåļŖö Ļ▓ĮĻ│╝ļź╝ Ļ░Ćņ¦ĆļŖö Ļ▓āņØ┤ Ļ░Ćņן ĒŖ╣ņ¦ĢņĀüņØ┤ļŗż. ņĄ£ņóģņĀüņ£╝ļĪ£ļŖö 76%ņØś ĒÖśņ×ÉņŚÉņä£ ĒåĄņ”ØņØä Ļ▓ĮĒŚśĒĢśņ¦Ćļ¦ī ņ¦äļŗ© ļŗ╣ņŗ£ ĒåĄņ”ØņØä ĒśĖņåīĒĢśļŖö Ļ▓ĮņÜ░ļŖö ļŗ©ņ¦Ć 54%ņŚÉ ļČłĻ│╝ĒĢśņśĆļŗż. ņØ┤ļŖö ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ņĪ░ĻĖ░ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś Ļ▓ĮņÜ░ Ļ░üĻ░ü 77%, 96%ņŚÉņä£ ņ¦äļŗ© ļŗ╣ņŗ£ ĒåĄņ”ØņØä ĒśĖņåīĒĢśļŖö Ļ▓āņŚÉ ļ╣äĻĄÉĒĢśļ®┤ ĻĘĖ ļ╣łļÅäļéś ņżæņ”ØļÅä ļ®┤ņŚÉņä£ ņāüļīĆņĀüņ£╝ļĪ£ Ļ│ĀĒåĄ ņŚåļŖö Ļ▓ĮĻ│╝ļź╝ ļ│┤ņØĖļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż[67].
ņÜöņĢĮĒĢśņ×Éļ®┤, ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝Ļ│╝ ņ×ÉņŚ░Ļ▓ĮĻ│╝ļź╝ ļŗ¼ļ”¼ĒĢśļŖö ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņØ┤ ņĪ┤ņ×¼ĒĢśĻ│Ā, ņØ┤ļōżņØĆ ļ░£ļ│æņŚ░ļĀ╣ņŚÉ ļö░ļØ╝ 2Ļ░Ćņ¦Ć ĒĢśņ£ä ņ¦æļŗ©ņ£╝ļĪ£ ļéśļłäņ¢┤ ņ¦Ćļ®░, ņØ┤ļōż ļśÉĒĢ£ Ļ░üĻĖ░ ļŗżļźĖ ņ×ÉņŚ░Ļ▓ĮĻ│╝ļź╝ ņĘ©ĒĢśļŖöļŹ░, ņĪ░ĻĖ░ĒśĢņØś Ļ▓ĮņÜ░ ņåīņĢäļéś ņ▓ŁņåīļģäĻĖ░ņŚÉ ņŗ£ņ×æĒĢśņŚ¼ ņŗ¼ĒĢ£ ĒåĄņ”ØņØ┤ ĒŖ╣ņ¦ĢņØ┤ņ¦Ćļ¦ī ļ╣äĻĄÉņĀü ņĘīņן ĻĖ░ļŖźņØ┤ ļ│┤ņĪ┤ļÉśĻ│Ā ņäØĒÜīĒÖöņØś ļ░£ņāØņØ┤ ļŖ”Ļ▓ī ļéśĒāĆļéśļ®░, ĒøäĻĖ░ĒśĢņØś Ļ▓ĮņÜ░ 50ļīĆ ņØ┤ĒøäņŚÉ ļ░£ļ│æĒĢśņ¦Ćļ¦ī ņāüļīĆņĀüņ£╝ļĪ£ ĒåĄņ”ØņØ┤ ņĀüņØĆ ņ¢æņāüņØä ļ│┤ņØĖļŗżĻ│Ā ĒĢĀ ņłś ņ׳ļŗż.
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝ņŚÉ ņśüĒ¢źņØä ļ»Ėņ╣śļŖö ņØĖņ×Éļōż
ņĢīņĮöņś¼
ņ¦ĆņåŹņĀüņØĖ ņĢīņĮöņś¼ ņäŁņĘ©Ļ░Ć ņØ┤ļ»Ė ņ¦äļŗ©ļÉ£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņśłĒøäņŚÉ ņ׳ņ¢┤ņä£ Ļ│äņåŹņĀüņØĖ ņĢģņśüĒ¢źņØä ņŻ╝ļ”¼ļØ╝ļŖö Ļ▓āņŚÉļŖö ņØ┤Ļ▓¼ņØ┤ ņŚåņ£╝ļ®░[85], ņłĀņØä ļüŖĻ▒░ļéś ņżäņØĖ ĒÖśņ×ÉņŚÉ ļ╣äĒĢ┤ ņ¦ĆņåŹņĀüņ£╝ļĪ£ ņłĀņØä ļ¦łņŗĀ ĒÖśņ×ÉļōżņŚÉņä£ ņé¼ļ¦ØļźĀĻ│╝ ņŗĀņ▓┤ņåÉņāüņ£╝ļĪ£ ņØĖĒĢ£ ņŗżņŚģļźĀņØ┤ 3ļ░░ņØ┤ņāü ļåÆņĢśļŗż[66]. ĒĢśņ¦Ćļ¦ī ĻĖłņŻ╝Ļ░Ć ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņ”ØņāüņØ┤ļéś ņןĻĖ░ņśłĒøäņŚÉ ņ¢╝ļ¦łļéś ĻĖŹņĀĢņĀüņØĖ ņśüĒ¢źņØä ņŻ╝ļŖöĻ░ĆņŚÉ ļīĆĒĢ┤ņä£ļŖö ņŚ░ĻĄ¼Ļ▓░Ļ│╝Ļ░Ć ļŗżņ¢æĒĢśļŗż. 32ļ¬ģņØś ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ņČöņĀüĻ┤Ćņ░░ĒĢśņśĆņØä ļĢī ĻĖłņŻ╝ ņØ┤ĒøäņŚÉļÅä ņĘīņןņØś ĻĖ░ļŖź ņĀĆĒĢśļŖö Ļ│äņåŹņĀüņ£╝ļĪ£ ņ¦äĒ¢ēĒĢśņ¦Ćļ¦ī ļŹö ļŖÉļ”¼Ļ│Ā Ļ▓ĮĒĢ£ Ļ▓ĮĻ│╝ļź╝ ļ│┤ņśĆĻ│Ā[86], ļŗżļźĖ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ĻĖłņŻ╝Ļ░Ć ĒåĄņ”ØņØ┤ļéś ņĘīņןņØś ņÖĖļČäļ╣äĻĖ░ļŖźņŚÉ ņ׳ņ¢┤ ļ¬ģļ░▒ĒĢ£ ĒśĖņĀäņØä ņŻ╝ņ¦Ć ļ¬╗ĒĢśņ¦Ćļ¦ī ņĘīņןņØś ļé┤ļČäļ╣äĻĖ░ļŖźņØä ĒśĖņĀäņŗ£ĒéżļŖö ĒÜ©Ļ│╝Ļ░Ć ņ׳ņŚłļŗż[68], ļśÉĒĢ£ ņä£ņ¢æņØś ņŚ¼ļ¤¼ ļ│┤Ļ│ĀņŚÉ ņØśĒĢśļ®┤ ĻĖłņŻ╝ļĪ£ 50-75%ņØś ĒÖśņ×ÉņŚÉņä£ ĒåĄņ”ØņØ┤ ņåīņŗżļÉśņŚłļŗż[73]. ņØ╝ļ│ĖņØś ļ│┤Ļ│ĀņŚÉ ļö░ļź┤ļ®┤ ņłĀņØä ļüŖĻ▒░ļéś ņżäņØĖ ĒÖśņ×ÉļŖö 60%ņŚÉņä£ ĒåĄņ”ØņØ┤ ĒśĖņĀäļÉśņŚłņ£╝ļéś ņ¦ĆņåŹņĀüņØĖ ņØīņŻ╝ļź╝ ĒĢ£ ĒÖśņ×ÉļŖö ĒåĄņ”ØņØś ņ×ÉņŚ░ ņåīņŗżņØ┤ 26% ļ░¢ņŚÉ ļÉśņ¦Ć ņĢŖņĢśļŗż[87]. ĒĢśņ¦Ćļ¦ī ņä£ņ¢æņØś ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ņ×Éļ░£ņĀüņØĖ ĻĖłņŻ╝ĻĄ░ņŚÉņä£ ļŗ©ņ¦Ć 52%ņŚÉņä£ļ¦ī ĒåĄņ”ØņØś ņåīņŗżņØ┤ ļ│┤ņśĆņ£╝ļéś, ņ¦ĆņåŹņĀüņØĖ ņØīņŻ╝ĻĄ░ņŚÉņä£ļÅä 37%ņŚÉņä£ļéś ņ×Éļ░£ņĀüņØĖ ĒåĄņ”ØņØś ņåīņŗżņØ┤ ņ׳ņŚłļŗż[68]. ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ņØ┤ļ»Ė ĒÖśņ×ÉĻ░Ć ļ¦ÉĻĖ░ņØś ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ ņÖĆ ņ׳ļŗżļ®┤ ņØīņŻ╝ņŚ¼ļČĆĻ░Ć ĒÖśņ×ÉņØś ĒåĄņ”ØņŚÉ ļ│ä ņśüĒ¢źņØä ņŻ╝ņ¦Ć ļ¬╗ĒĢśņśĆļŗż[66]. ļö░ļØ╝ņä£ ĻĖłņŻ╝Ļ░Ć ļ¬©ļōĀ ĒÖśņ×ÉņŚÉņä£ ĒåĄņ”ØņØś ņåīņŗżņØ┤ļéś ņĘīņן ĻĖ░ļŖźņØś ĒśĖņĀäņØä ļ│┤ņןĒĢ┤ ņŻ╝ņ¦ĆļŖö ļ¬╗ĒĢśņ¦Ćļ¦ī, ĻĖłņŻ╝ļź╝ ĒĢśļ®┤ ņĀüņ¢┤ļÅä ļ░ś ņĀĢļÅäņØś ĒÖśņ×ÉņŚÉņä£ļŖö ĒåĄņ”ØņØś ĒśĖņĀäņØä ĻĖ░ļīĆĒĢĀ ņłś ņ׳Ļ│Ā, ņĘīņן ĻĖ░ļŖźņĀĆĒĢśņØś ņ¦ĆņŚ░ņØä Ļ░ĆņĀĖņś¼ ņłś ņ׳ņ£╝ļ®░ ņ¦ĆņåŹņĀüņØĖ ņØīņŻ╝Ļ░Ć ļ¦īņä▒ņĘīņןņŚ╝ņØś ņśłĒøäņŚÉ ņĢģņśüĒ¢źņØä ņŻ╝ļŖö Ļ▓āņØĆ ļ¬ģļ░▒ĒĢśļ»ĆļĪ£ ļ░śļō£ņŗ£ ļ¬©ļōĀ ĒÖśņ×ÉņŚÉĻ▓ī ĻĖłņŻ╝ļź╝ ĻČīĒĢ┤ņĢ╝ ĒĢĀ Ļ▓āņØ┤ļŗż.
ĒØĪņŚ░
ĒØĪņŚ░ņØĆ ņØ┤ņĀ£ ļ¦īņä▒ņĘīņןņŚ╝ ļ░£ņāØ ļ░Å ņ¦äĒ¢ēņØś ļÅģļ”ĮņĀü ņ£äĒŚśņØĖņ×ÉļĪ£ ņāØĻ░üĒĢ£ļŗż[88]. ļīĆļČĆļČäņØś ļ¦īņä▒ ņØīņŻ╝ņ×ÉļōżņØ┤ ļŗ┤ļ░░ļź╝ Ēö╝ņÜ░ļ»ĆļĪ£ ņĢīņĮöņś¼Ļ│╝ ĒØĪņŚ░ņØś ņśüĒ¢źņØä ļö░ļĪ£ ļČäļ”¼ĒĢśļŖö Ļ▓āņŚÉ ņ¢┤ļĀżņøĆņØ┤ ņ׳ņŚłņ¦Ćļ¦ī, Talamini ļō▒ņØĆ ņĢīņĮöņś¼ņä▒ ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ĒØĪņŚ░ņØ┤ ļÅģļ”ĮņĀüņØĖ ņ£äĒŚśņÜöņØĖņ×äņØä ņ”Øļ¬ģĒĢśņśĆĻ│Ā[89], ļ╣ä ņĢīņĮöņś¼ņä▒ ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×Éļź╝ ļīĆņāüņ£╝ļĪ£ ĒĢ£ ņŚ░ĻĄ¼ņŚÉņä£ļŖö ņäØĒÜīĒÖöņÖĆ ļŗ╣ļć©ļ│æņØś ļ░£ĒśäņØä ņ¦łļ│æņØś ņ¦äĒ¢ēņ£╝ļĪ£ ĒīÉļŗ©ĒĢśņśĆņØä ļĢī ĒØĪņŚ░ņ×ÉļōżņŚÉĻ▓īņä£ ļåÆņØĆ ņ£äĒŚśļÅäļź╝ ļ│┤ņŚ¼ ĒØĪņŚ░ņØ┤ ļ¦īņä▒ņĘīņןņŚ╝ ņ¦łļ│æĻ▓ĮĻ│╝ļź╝ ļŹöņÜ▒ ļ╣Āļź┤Ļ▓ī ņ¦äĒ¢ēņŗ£Ēé┤ņØä ļ│┤ņŚ¼ņŻ╝ņŚłļŗż[90]. ļśÉĒĢ£ ĒØĪņŚ░ņØĆ ļŗłņĮöĒŗ┤ ņä▒ļČäņØä ĒåĄĒĢ┤ ņĘīņןĒÜ©ņåī ļČäļ╣ä Ēī©Ēä┤ņŚÉ ņśüĒ¢źņØä ņżä ļ┐É ņĢäļŗłļØ╝ ņŚ╝ņ”Øļ░śņØæņØä ņĢ╝ĻĖ░ĒĢśĻ│Ā ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņä▒ļČäļōżņØä ĒåĄĒĢ┤ ļ░£ņĢöĒÜ©Ļ│╝ļź╝ ļéśĒāĆļéĖļŗż[91]. Ļ▓░ĻĄŁ ĒØĪņŚ░ņØ┤ ņĢīņĮöņś¼ļĪ£ ņĢ╝ĻĖ░ļÉ£ ņĘīņן ņåÉņāüņØä ļŹöņÜ▒ ņĢģĒÖöņŗ£ĒéżĻ│Ā ļ¦īņä▒ņĘīņןņŚ╝ ļ░£ņāØĻ│╝ ņ¦äĒ¢ēņØä Ļ░ĆņåŹĒÖöĒĢĀ ļ┐É ņĢäļŗłļØ╝ ņĘīņןņĢö ļ░£ņāØņŚÉļÅä ĻĖ░ņŚ¼ĒĢśļŖö Ļ▓āņØĆ ļČäļ¬ģĒĢśļ»ĆļĪ£ ņØīņŻ╝ņÖĆ ļ¦łņ░¼Ļ░Ćņ¦ĆļĪ£ ļ¬©ļōĀ ĒÖśņ×ÉļōżņŚÉĻ▓ī ĻĖłņŚ░ņØä ĻČīĒĢ┤ņĢ╝ ĒĢĀ Ļ▓āņØ┤ļŗż.
ņ╣śļŻī
ļé┤ņŗ£Ļ▓Į ņ╣śļŻīļź╝ ĒżĒĢ©ĒĢ£ ļ│┤ņĪ┤ņĀüņØĖ ņ╣śļŻīļéś ņłśņłĀ ņ╣śļŻīĻ░Ć ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ×ÉņŚ░Ļ▓ĮĻ│╝ļéś ņśłĒøäņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØĆ ļ»Ėļ»ĖĒĢśļŗż. ņØ┤ļŖö ļīĆļČĆļČäņØś ļé┤ņŗ£Ļ▓Į ļ░Å ņłśņłĀ ņ╣śļŻīĻ░Ć ņØ┤ļ»Ė ņ¦äĒ¢ēļÉ£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉļōżņØś ĒåĄņ”Ø ņĪ░ņĀł ņĖĪļ®┤ņŚÉņä£ļ¦ī ļŗżļŻ©ņ¢┤ņĪīĻĖ░ ļĢīļ¼ĖņØ┤ļ®░, ļŗ©ņ¦Ć ĒåĄņ”ØņØś ņĪ░ņĀłņØ┤ļØ╝ļŖö Ļ┤ĆņĀÉņŚÉņä£ļ¦ī ļ░öļØ╝ļ│Ėļŗżļ®┤ ļé┤ņŗ£Ļ▓Į ņ╣śļŻīņÖĆ ņłśņłĀ ņ╣śļŻī ņä▒ņĀüņŚÉ ļīĆĒĢ┤ņä£ļŖö ļŗżņ¢æĒĢ£ ņŚ░ĻĄ¼Ļ▓░Ļ│╝Ļ░Ć ņ׳ļŗż. Ļ░Ćņן ņĄ£ĻĘ╝Ļ╣īņ¦ĆņØś ņĀäĒ¢źņĀü ņŚ░ĻĄ¼ņä▒Ļ│╝ļōżņØä ļ╣äĻĄÉĒĢ£ļŗżļ®┤ ņłśņłĀ ņ╣śļŻīĻ░Ć ĒåĄņ”ØņØś ņןĻĖ░ ņĪ░ņĀłņŚÉ ņ׳ņ¢┤ņä£ ļé┤ņŗ£Ļ▓Į ņ╣śļŻīļ│┤ļŗż ņÜ░ņ£äņŚÉ ņ׳ļŖöĻ▓āņ£╝ļĪ£ ļ│┤Ļ│ĀļÉ£ļŗż[92-94].
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņśłĒøä
ĒĢ®ļ│æņ”Ø
ļ¦īņä▒ņĘīņןņŚ╝ņØś ĒĢ®ļ│æņ”ØņØĆ ņĘīņןņØś Ļ░Ćņä▒ļéŁņóģĻ│╝ ļåŹņ¢æ, ņ┤Øļŗ┤Ļ┤Ć ļ░Å ņŗŁņØ┤ņ¦ĆņןņØś Ēśæņ░®, ļ│ĄņłśņÖĆ ļŖæļ¦ēņé╝ņČ£, ļ╣äņןņĀĢļ¦ź ĒśłņĀäņ”ØņŚÉ ņØśĒĢ£ ļ¼Ėļ¦źņĢĢĒĢŁņ¦äņ”Ø ļō▒ ļ¦żņÜ░ ļŗżņ¢æĒĢśĻ│Ā, ņØ┤ļ¤¼ĒĢ£ ļ¬©ļōĀ ĒĢ®ļ│æņ”ØļōżņØĆ ĒÖśņ×ÉņØś ņśłĒøäņŚÉ ļéśņü£ ņśüĒ¢źņØä ņŻ╝ļŖö Ļ▓āņØĆ ĒŗĆļ”╝ņØ┤ ņŚåļŗż. ĒĢśņ¦Ćļ¦ī ņØ┤ņŚÉ ļīĆĒĢ£ ļīĆĻĘ£ļ¬© ņ×äņāü ņŚ░ĻĄ¼Ļ░Ć ņŚåĻĖ░ ļĢīļ¼ĖņŚÉ Ēśäņ×¼Ļ╣īņ¦Ć ļ¦īņä▒ņĘīņןņŚ╝ Ļ▓ĮĻ│╝ņŚÉ ņĀĢĒÖĢĒ׳ ņ¢┤ļ¢ż ņśüĒ¢źņØä ņŻ╝ļŖöņ¦ĆļŖö ļČłļ¬ģĒÖĢĒĢśļŗż[73].
ņĘīņןņĢö
ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ņĘīņןņĢöņØś ļ░£ņāØļźĀņØĆ 1.4-2.7%Ļ╣īņ¦Ć ļŗżņ¢æĒĢśĻ▓ī ļ│┤Ļ│ĀļÉśņŚłļŗż[68,69,73]. ĒĢ£ ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ņŚÉ ņØśĒĢśļ®┤ ņĘīņןņĢöņØś ļłäņĀüļ░£ņāØņ£äĒŚśļÅäļŖö ņĘīņןņŚ╝ ņ¦äļŗ© 2ļģäņ¦ĖļČĆĒä░ ĒÖĢņŗżĒ׳ ņ”ØĻ░ĆĒĢśĻĖ░ ņŗ£ņ×æĒĢśļ®░ ņ¦äļŗ© Ēøä 10ļģä, 20ļģäņ¦ĖņØś ņ£äĒŚśļÅäļŖö Ļ░üĻ░ü 1.8%ņÖĆ 4%ļĪ£ ņĢīļĀżņĀĖ ņ׳ļŗż[95]. ļö░ļØ╝ņä£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ņĘīņןņĢöņØś ņ£äĒŚśņØĆ ļČäļ¬ģĒ׳ ņ”ØĻ░ĆĒĢśļ®░ ļ¦īņä▒ņĘīņןņŚ╝ņØĆ ņĀäņĢö ļŗ©Ļ│äļĪ£ ņāØĻ░üĒĢĀ ņłś ņ׳ļŗż. ĒĢśņ¦Ćļ¦ī ņĢäņ¦üĻ╣īņ¦ĆļÅä ņĘīņןņĢöņØś ņ¦äļŗ© ņ×Éņ▓┤Ļ░Ć ņēĮņ¦Ć ņĢŖĻ│Ā, ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ņĘīņןņĢöņØś ņĪ░ĻĖ░ņ¦äļŗ©ņØä ņ£äĒĢ£ ļ░®ļ▓ĢņØĆ ņĀĢļ”ĮļÉśņ¢┤ ņ׳ņ¦Ć ņĢŖļŗż.
ņĘīņןņĢö ņØ┤ņÖĖņØś ļŗżļźĖ ņĢöņØś ļ░£ņāØ
ņØīņŻ╝Ļ░Ć ļ¦īņä▒ņĘīņןņŚ╝ņØś ņŻ╝ļÉ£ ņøÉņØĖņØ┤Ļ│Ā, ņØīņŻ╝ņ×É ņżæņŚÉ ĒØĪņŚ░ņ×ÉĻ░Ć ļ¦Äņ£╝ļ»ĆļĪ£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×É ņżæ ĻĄ¼Ļ░Ģ, ņØĖĒøä ļ░Å ĒÅÉņĢö ļō▒ ņāüļČĆĒśĖĒØĪĻĖ░ ņĢöņØś ļ░£ņāØļ╣łļÅäĻ░Ć ļåÆļŗż. ņØ┤ļōż ņĢöņØä ĒżĒĢ©ĒĢśņŚ¼ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņŚÉņä£ ņĘīņן ņÖĖ ņĢöņØś ļ░£ņāØ ļ╣łļÅäļŖö 3.9-12.5%ļĪ£ ļåÆĻ▓ī ļ│┤Ļ│ĀļÉ£ļŗż[73].
ņé¼ļ¦ØļźĀĻ│╝ ņé¼ļ¦Ø ņøÉņØĖ
ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņé¼ļ¦ØļźĀņŚÉ ļīĆĒĢ£ ņ×ÉļŻī ĒĢ┤ņäØņØĆ ņ×äņāü ņŚ░ĻĄ¼ņŚÉ ļö░ļØ╝ ņøÉņØĖĻ│╝ Ļ┤Ćņ░░ ĻĖ░Ļ░äņØś ņ░©ņØ┤Ļ░Ć ļŗżņ¢æĒĢśļ»ĆļĪ£ ņ¢┤ļĀĄļŗż. ļ╣äĻĄÉņĀü ļ╣äņŖĘĒĢ£ Ļ┤Ćņ░░ĻĖ░Ļ░ä(6.3-9.8ļģä)ņØä Ļ░Ćņ¦ĆļŖö 3Ļ░£ņØś ņ×äņāü ņĮöĒśĖĒŖĖ ņŚ░ĻĄ¼ņŚÉ ņØśĒĢśļ®┤ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ņØ╝ļ░ś ņé¼ļ¦ØļźĀņØĆ 28.8-35%ņØ┤ņ¦Ćļ¦ī, ņØ┤ ņżæ ņĘīņןņĢöņØä ĒżĒĢ©ĒĢśņŚ¼ ļ¦īņä▒ņĘīņןņŚ╝ņØś ĒĢ®ļ│æņ”ØĻ│╝ ņŚ░Ļ┤ĆļÉ£ ņé¼ļ¦ØļźĀņØĆ ļŗ©ņ¦Ć 12.8-19.8%ņśĆļŗż[68,69,87]. ļ¦ÄņØĆ ĒÖśņ×ÉļōżņØś ņé¼ļ¦ØņøÉņØĖņØĆ ņĘīņן ņÖĖ ņ¦łĒÖś, ņ”ē ņĘīņןņØä ņĀ£ņÖĖĒĢ£ ļŗżļźĖ ņןĻĖ░ņĢö, ņŗ¼ĒśłĻ┤ĆĻ│ä ņ¦łĒÖś, ņżæņ”Ø Ļ░ÉņŚ╝ņ”Ø ļō▒Ļ│╝ Ļ┤ĆĻ│äĻ░Ć ņ׳ņŚłļŗż[17,69]. ļśÉĒĢ£ ņøÉņØĖņŚÉ ļö░ļØ╝ ņé¼ļ¦ØļźĀņØś ņ░©ņØ┤Ļ░Ć ļéśĒāĆļéśļŖöļŹ░, ņ”ē ĒŖ╣ļ░£ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉ ļ╣äĒĢ┤ ņĢīņĮöņś¼ņä▒ ļ¦īņä▒ņĘīņןņŚ╝ņŚÉņä£ ņé¼ļ¦ØļźĀņØ┤ ļŹö ļåÆļŗż[67,68].
ļ¦īņä▒ņĘīņןņŚ╝ņØś ņé¼ļ¦ØļźĀņŚÉ ļīĆĒĢ£ ļīĆĻĘ£ļ¬© ļŗżĻĖ░Ļ┤Ć ņŚ░ĻĄ¼ņŚÉņä£ļŖö ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉĻ░Ć ņĘīņןņŚ╝ņØ┤ ņŚåļŖö ņé¼ļ×īļ│┤ļŗż 3.6ļ░░ņØś ņé¼ļ¦ØļźĀņØä ļ│┤ņśĆļŗż. ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś 10ļģä, 20ļģä ņāØņĪ┤ņ£©ņØĆ Ļ░üĻ░ü 70%, 45%ļĪ£ ņĀĢņāü ļīĆņĪ░ĻĄ░ņØś 93%, 65%ļ│┤ļŗż ļé«ņĢśļŗż. ļŗżļ│Ćļ¤ē ļČäņäØņŚÉņä£ ļÅģļ”ĮņĀüņØĖ ņé¼ļ¦ØņØś ņŻ╝ņÜö ņ£äĒŚśņÜöņåīļĪ£ļŖö ņ¦äļŗ© ļŗ╣ņŗ£ņØś ļ¦ÄņØĆ ļéśņØ┤(ĻĄÉņ░©ļ╣ä 2.3-6.3), ņ¦ĆņåŹņĀüņØĖ ņØīņŻ╝(1.6), ĒØĪņŚ░(1.4), Ļ░äĻ▓Įļ│ĆņØś ļÅÖļ░ś(2.5)ņØ┤ ņ׳ņŚłĻ│Ā, ņä▒ļ│äņØ┤ļéś ņłśņłĀņŚ¼ļČĆļŖö ņśłĒøäņÖĆ ļ¼┤Ļ┤ĆĒĢśņśĆļŗż[95].
Ļ▓░ ļĪĀ
ļ¦īņä▒ņĘīņןņŚ╝ņØĆ TIGAR-O ļČäļźśĒæ£ņŚÉņä£ ņĀ£ņŗ£ĒĢ£ ļ░öņÖĆ Ļ░ÖņØ┤ ļ¦żņÜ░ ļŗżņ¢æĒĢ£ ņøÉņØĖ ņØĖņ×ÉņŚÉ ņØśĒĢ┤ ļ░£ļ│æļÉśļŖö Ļ▓āņ£╝ļĪ£ ņĢīļĀżņĀĖ ņ׳ņ£╝ļéś ņøÉņØĖĻ│╝ ņ¦łļ│æņØś ņāüĻ┤ĆĻ┤ĆĻ│äņØś ņ×¼Ēśäņä▒ņØĆ ĻĘĖļŗżņ¦Ć ļåÆņ¦Ć ņĢŖņĢä Ļ░£ņØĖņĀü ĒÖśĻ▓Į ļ░Å ņ£ĀņĀäņĀü Ļ░Éņłśņä▒ņØ┤ ļ│æņØś ļ░£ņāØņŚÉ Ēü¼Ļ▓ī Ļ┤ĆņŚ¼ĒĢśļŖö Ļ▓āņ£╝ļĪ£ ņČöņĖĪļÉśĻ│Ā ņ׳ļŗż. ņØ┤ļ¤¼ĒĢ£ Ļ░£ņØĖņĀü Ļ░Éņłśņä▒ņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼ļĪ£ ņ£ĀņĀäņ×Éļ│ĆņØ┤ ļō▒ņŚÉ ļīĆĒĢ£ ņŚ░ĻĄ¼Ļ░Ć ĒÖ£ļ░£ĒĢ┤ņ¦ĆĻ│Ā ņ׳ņ£╝ļéś ņĢäņ¦üĻ╣īņ¦ĆļŖö ļ░£Ļ▓¼ļÉ£ ņ£ĀņĀäņ×É ļÅīņŚ░ļ│ĆņØ┤ņÖĆ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ņāØņØś ņāüĻ┤ĆĻ┤ĆĻ│äņŚÉ ļīĆĒĢ£ ņĀĢĒÖĢĒĢ£ ĻĖ░ņĀäņØ┤ ļ░ØĒśĆņ¦Ćņ¦Ć ņĢŖņĢśļŗż. ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ│æļ”¼ ņåīĻ▓¼ņØĖ ņĘīņäĀļ░®ņäĖĒżņØś ņåīņŗż, ņŚ╝ņ”ØņäĖĒżņØś ņ╣©ņ£ż, ņä¼ņ£ĀĒÖöņŚÉ ļö░ļźĖ ĒĢ┤ļČĆĒĢÖņĀü ļ│ĆņØ┤Ļ░Ć ņ┤łļל ļÉśļŖö ĻĖ░ņĀäņØĆ ņĢäņ¦ü ļ¬ģĒÖĢĒĢśņ¦Ć ņĢŖņ£╝ļéś ņĄ£ĻĘ╝ ņĘīņן ņä¼ņ£ĀĒÖöņØś ĒĢĄņŗ¼ņĀüņØĖ ņäĖĒżļĪ£ņä£ ņĘīņä▒ņāüņäĖĒżņØś ļČäļ”¼ ļ░░ņ¢æņØ┤ Ļ░ĆļŖźĒĢ┤ņ¦ĆĻ│Ā ņäĖĒżņāØļ¼╝ĒĢÖņĀü ĻĖ░ļŖźņØ┤ ļ░ØĒśĆņ¦ÉņŚÉ ļö░ļØ╝ ņä¼ņ£ĀĒÖö ĻĖ░ņĀäņŚÉ ļīĆĒĢśņŚ¼ļŖö ļ│┤ļŗż Ļ╣ŖņØĆ ņØ┤ĒĢ┤Ļ░Ć ņ¦äĒ¢ēļÉśĻ│Ā ņ׳ļŗż. ĻĘĖļ¤¼ļéś ņĢäņ¦ü ņŚ¼ļ¤¼ Ļ░Ćņ¦Ć ņøÉņØĖņØĖņ×ÉĻ░Ć ņ┤łĻĖ░ņŚÉ ņĘīņäĀļ░® ņäĖĒżņØś ņåīņŗż ļ░Å ņĘīņä▒ņāüņäĖĒżņØś ĒÖ£ņä▒ĒÖöļź╝ ņ¢┤ļ¢╗Ļ▓ī ņ£ĀļÅäĒĢśļŖöņ¦ĆņŚÉ ļīĆĒĢ£ ņØ┤ĒĢ┤ļŖö ļČĆņĪ▒ĒĢ£ ļŗ©Ļ│äņØ┤ļŗż. Ē¢źĒøä ņ×äņāüņĀüņ£╝ļĪ£ ĒĢ®ļŗ╣ĒĢ£ ļÅÖļ¼╝ļ¬©ļŹĖņØś Ļ░£ļ░£, ļČäņ×É ņ£ĀņĀäĒĢÖņĀü ĻĖ░ņłĀņØś ļ░£ņĀä, ļ¦īņä▒ņĘīņןņŚ╝ņØś ņ¦äļŗ©ļ░®ļ▓ĢņØś Ļ░£ņäĀņØä ĒåĄĒĢśņŚ¼, ļ¦īņä▒ņĘīņןņŚ╝ņØś ņĀä ņ×äņāüļŗ©Ļ│äņŚÉņä£ņØś ņĪ░ĻĖ░ņ¦äļŗ© ļō▒ņØ┤ Ļ░ĆļŖźĒĢśĻ▓ī ļÉ£ļŗżļ®┤ ļ¦īņä▒ņĘīņןņŚ╝ņØś ļ░£ņāØ ĻĖ░ņĀäņŚÉ ļīĆĒĢ£ ņØ┤ĒĢ┤Ļ░Ć ņ¦äņĀäļÉĀ Ļ▓āņ£╝ļĪ£ ņāØĻ░üļÉ£ļŗż.
ņ¦äĒ¢ēļÉ£ ļ¦īņä▒ņĘīņןņŚ╝ņØś Ļ▓ĮņÜ░ ņśłĒøäĻ░Ć ņāüļŗ╣Ē׳ ļČłļ¤ēĒĢśĻ│Ā ņ╣śļŻīĻ░Ć ņ×ÉņŚ░Ļ▓ĮĻ│╝ņŚÉ ļ»Ėņ╣śļŖö ņśüĒ¢źņØĆ ļ»Ėļ»ĖĒĢśļ»ĆļĪ£ ņĪ░ĻĖ░ņŚÉ ļ¦īņä▒ņĘīņןņŚ╝ņØä ņ¦äļŗ©ĒĢśļŖö Ļ▓āņØ┤ ĒĢäņÜöĒĢśļŗż. ļ╣äļĪØ ļ¦īņä▒ņĘīņןņŚ╝ņØś ņĀĢņØśĻ░Ć ļ╣äĻ░ĆņŚŁņä▒ņØä ļé┤ĒżĒĢśņ¦Ćļ¦ī ņ×ÉņŚ░Ļ▓ĮĻ│╝ ņżæ Ļ░ĆņŚŁņĀüņØĖ ļŗ©Ļ│äĻ░Ć ļČäļ¬ģĒ׳ ņĪ┤ņ×¼ĒĢśļ»ĆļĪ£ ņØ┤ļź╝ ņ░ŠņĢäļé┤ņ¢┤ ļ╣äĻ░ĆņŚŁņĀüņØĖ ļ¦īņä▒ņĘīņןņŚ╝ņ£╝ļĪ£ņØś ņ¦äĒ¢ē ļ░Å ņĘīņןņĢöņØś ļ░£ļ│æņØä ņśłļ░®ĒĢśļŖö Ļ▓āņØ┤ ņĢ×ņ£╝ļĪ£ ņ¦äĒ¢ēļÉĀ ņŚ░ĻĄ¼ņŚÉ Ļ░Ćņן ņżæņÜöĒĢ£ ĒÖöļæÉĻ░Ć ļÉĀ Ļ▓āņØ┤ļŗż.
ņØ┤ļ»Ė ņ¦äļŗ©ļÉ£ ļ¦īņä▒ņĘīņןņŚ╝ ĒÖśņ×ÉņØś ĒåĄņ”ØņØ┤ļéś ļé┤ŃåŹņÖĖļČäļ╣ä ĻĖ░ļŖź ļČĆņĀäņØś ņ╣śļŻīļź╝ ņĀĢĒśĢĒÖöĒĢśļŖö Ļ▓āņØĆ ļČłĻ░ĆļŖźĒĢśņ¦Ćļ¦ī ņØ┤ņŚÉ ļīĆĒĢ£ ļ¬ć Ļ░Ćņ¦Ć ņŚ░ĻĄ¼ņä▒Ļ│╝ļōżņØĆ ņŗżņĀ£ ņ¦äļŻīņŚÉ ņ׳ņ¢┤ ĒĢśļéśņØś ņ¦Ćņ╣©ņØä ņżä ņłś ņ׳ņ£╝ļ®░ Ļ░Ćņן ļ©╝ņĀĆ ĻĖłņŚ░, ĻĖłņŻ╝ļź╝ ĻČīĒĢśļŖö Ļ▓āņØ┤ ņżæņÜöĒĢśļŗż.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






