


서 론
췌장의 신경내분비종양은 내분비분화의 특성을 갖는 상피종양이다. 췌장의 관샘암종과 비교하여 비록 전이가 있을지라도 예후가 좋으므로 정확한 진단과 적절한 치료가 중요하다. 빈도는 전체 인구 약 10만 명당 1명 이하로 드물며, 전체 췌장종양의 1~2%를 차지하는 드문 종양이다[1]. 그러나 부검자료에 따르면 10%에 달한다고 하며[2] 영상의학의 발달로 우연히 발견하는 경우도 증가하는 것으로 보아[3] 임상적으로 발견하지 못하는 신경내분비종양은 더욱 많으리라고 본다. 임상적으로 기능성일 수도, 비기능성일 수도 있는데 일반적으로 약 30~40%가 비기능성이다.
췌장은 외분비세포와 내분비세포가 공존하는 장기이다. 외분비세포로 구성된 샘 사이에서 내분비세포는 섬처럼 모여서 위치하는데 이것이 1869년 Paul Langerhans가 처음 발견하고 기술한 “islets of Langerhans”이다(Fig. 1). 한 개의 섬은 약 3,000개의 세포로 구성되어 있으며 구성세포는 매우 다양하다. 인슐린을 분비하는 베타세포가 약 반수를 차지하며 글루카곤을 생성하는 알파세포가 약 1/3을 차지하고 소마토스타틴을 생성하는 델타세포가 약 10%를 차지하며 나머지 vasoactivie intestinal polypeptide (VIP), pancreatic polypeptide (PP), substance P/세로토닌 등으로 구성된다. 가스트린을 생성하는 G세포는 태생기에 있으나 성인에서는 없다.
신경내분비세포는 정상 장의 내분비세포와 유사한 세포로서 면역염색을 시행하면 일반적인 신경내분비 표지자인 chromogranin A와 synaptophysin 등에 강양성으로 염색되고 부위에 따라 특이 호르몬을 함유하고 있다. 이러한 신경내분비세포로 이루어진 분화가 좋은 종양이 신경내분비종양이며 심한 핵 비정형 및 괴사 및 다수의 세포분열이 관찰되는 경우 신경내분비암종이라고 할 수 있다.
정의 및 분류
정의
신경내분비종양은 종양세포의 특징적인 형태와 배열양상이 통상적인 암종의 그것과 유사하다고 하여 유암종(carcinoid tumor)이라고 불리어 왔으며, 생물학적 성상에 따라 비정형 유암종, 악성 유암종 등으로 구분하였다. 그러나 종양이라는 접미어는 양성 질환에 붙이는 것으로서 신경내분비종양의 생물학적 특성에 견주어 본다면 잘못된 용어이다. 신경내분비라는 기본 개념은 발생 중 신경능으로부터 기원한 세포라는 개념을 갖고 있었다. 그러나 실제로 종양세포는 상피세포종양이며 내배엽기원 세포의 특성을 보임에 따라 이전 국제보건기구(World Health Organization, WHO) 2000년 분류에서는 내분비종양이라는 용어를 도입하여 사용하여 고분화내분비종양, 고분화내분비암종, 저분화내분비암종 및 소세포암종 등으로 구분하였다. WHO 2004년에는 생물학적 성상에 따라 양성 내분비종양, 예측하기 어려운 행동양식의 내분비종양, 악성 내분비종양 등으로 구분하였으나 여전히 내분비종양이라고 지칭하였다. 그러나 WHO 2010년 분류[1]에서는 췌장의 신경내분비종양도 내분비기원 상피세포의 특징과 신경세포의 특징을 동시에 갖고 있으므로 위장관의 신경내분비기원의 종양과 같은 용어를 적용하여 췌장에서도 내분비종양이라는 용어 대신 신경내분비종양이라고 통일하여 부르게 되었다[4]. 즉 모든 장기에서 발생하는 신경내분비세포기원 종양을 통칭하여 신경내분비종양으로 부르고 고분화 신경내분비종양(well differentiated neuroendocrine tumor), 고분화 신경내분비암종(well differentiated neuroendocrine carcinoma), 저분화 신경내분비암종(poorly differentiated neuroendocrine carcinoma)의 세 영역으로 구분하고(Table 1) 등급과 장기에 따른 병기를 구분하여 산정하였다[5].
분화
고분화와 저분화로 나뉘며 분화정도는 기원세포의 성질을 얼마나 잘 표현하느냐의 정도에 따라 다르다. 고분화 신경내분비종양은 종양세포가 기관형성을 하듯이 소, 세포주 및 대뇌주름을 보는 듯한 배열을 하며 세포질 내 다수의 신경분비과립을 함유하고 있어 chromogranin A나 synaptophysin 등 면역염색 표지자에 양성이다. 반면 저분화 신경내분비종양은 판상 또는 아무런 배열양상이 없이 광범위하게 증식하며 핵은 불규칙하고 세포질의 과립성은 떨어지게 된다.
등급
등급 기준은 형태학적 기준과 세포분열수 또는 Ki67 세포증식지수에 따라 적용을 한다. 일부 학자는 췌장의 신경내분비종양에서도 폐나 가슴샘에서와 같이 세포분열수와 함께 괴사유무를 참고하여 등급을 나누기도 하였으나[6,7] 유럽신경내분비종양연합(European Neuroendocrine Tumor Society, ENETS) [8] 및 WHO 2010년 분류[1]에서는 10개의 고배율시야당 세포분열수로 기준을 정하였다(Table 2). 즉 G1은 세포분열수가 10개의 고배율시야당 2개 이하 또는 Ki67 지수가 2% 이하일 때, G2는 세포분열수가 10개의 고배율시야당 20개 이하 또는 Ki67 지수가 20% 이하일 때, G3는 세포분열수 및 Ki67지수가 G2보다 높을 때로 정한다.
병리소견
육안소견
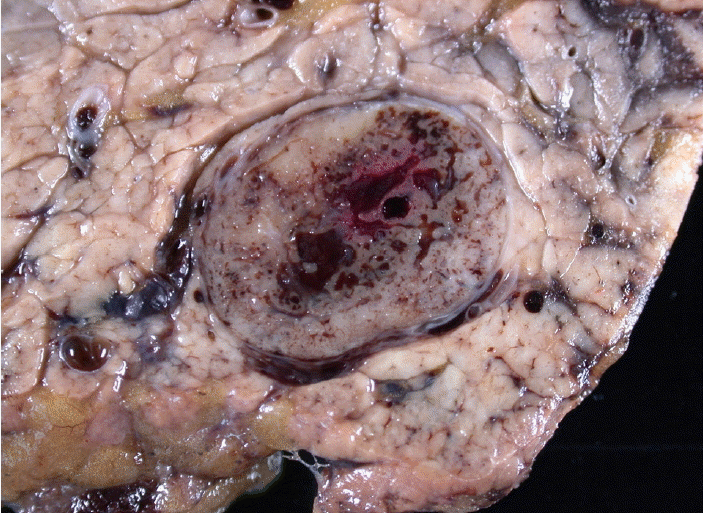
췌장의 어느 부위든 생길 수 있으나 췌장 두부에서 더 흔하며 고등급 신경내분비암종은 두부에서 더 흔하다. 일반적으로 췌장의 신경내분비종양은 경계가 좋으며 때로 섬유조직으로 이루어진 거짓피막으로 주변과 구별할 수 있다. 신경내분비암종은 대개 4 cm 이상이다. 단단하고 회백색의 종괴로서 주변과 경계가 뚜렷하지 않다. 단독으로 발생하고 황백색 또는 분홍색을 띤 갈색으로 나타난다. 경도는 생선살같이 연하거나 또는 단단할 수 있으며 출혈 및 괴사를 동반할 수 있다(Fig. 2). 두부에 발생한 경우 주췌관을 밀어 협착을 초래할 수 있으나 직접 침윤하지는 않는다. 드물게 낭으로 나타나 점액성 낭종양을 비롯한 다른 낭 종양과 감별이 필요할 수 있다. 심한 섬유화를 동반할 경우는 경계가 불분명하고 백색을 띠는 매우 단단한 종괴로 나타나서 관암종과의 감별이 필요할 수 있다. 종양세포의 세포질 내 다량의 리포훅신 색소를 함유한 경우 췌장의 흑색 내분비종양(pigmented black, PEN)이라고 부르며, 세포질 내 다량의 지방을 함유하여 부신피질종양과 유사하게 황색 내분비종양(lipid-rich yellow, PEN)으로 나타날 수 있다. 비기능성이면서 종양의 직경이 0.5 cm 미만인 작은 종양을 가리켜 췌장의 미소신경내분비종(neuroendocrine microadenom)이라고 부른다.
현미경 소견
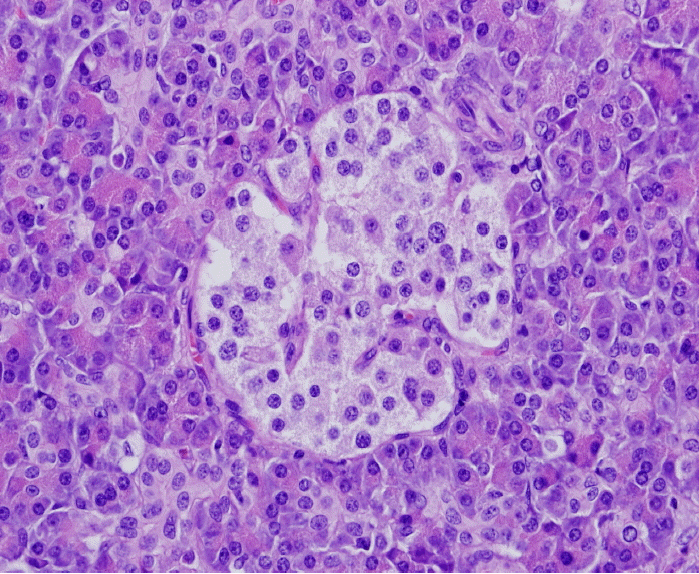
신경내분비종양은 개개의 세포는 균일한데 반해 배열양상은 특징적으로 기관을 형성하는 듯 한 모양(organoid), 즉 소(nest)형성, 세포주모양, 샘모양, 뇌주름모양, 관소엽모양, 거짓로젯 배열 등 다양한 모습을 취한다(Fig. 3). 개별 세포는 중심에 위치한 핵은 미세한 과립모양이고 핵소체가 작지만 뚜렷하며 세포질은 과립성이다. 염색질은 특징적으로 거칠게 뭉쳐서 마치 크림스프에 후추가루를 뿌려놓은 모양과 유사하다고 하여 “salt and pepper”양상이라고 부른다(Fig. 4).
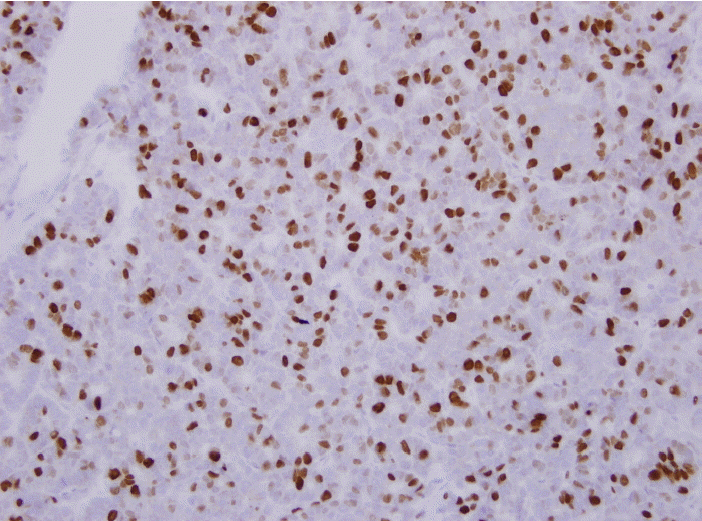
세포분열수는 10개 고배율시야당 20개 미만과 20개 이상으로 등급 결정에 매우 중요한 지표이다(Fig. 4). 이때 세포분열수의 산정은 최소 50개의 고배율시야를 세어 평균값을 구해야 한다. Ki67 세포증식지수도 2%와 20%를 기준으로 등급을 나누는 중요한 기준이다(Fig. 5). Ki67지수 산정은 가장 세포증식이 활발한 부위에서 500~2,000개의 세포를 세어 구하도록 한다. 세포분열수는 등급기준이 장기마다 다소 차이가 있고 생검조직은 여러 개의 시야를 관찰하기에 제한적이며 판독의사에 따라 차이가 있을 수 있으므로 병리보고서 내에서는 등급과 함께 실제 10개의 고배율시야당 세포분열수가 몇 개인지 구체적으로 명시하는 것이 바람직하다.
괴사가 국소적으로 있을 수 있으나 대부분 여드름모양의 중심괴사가 일반적이며 광범위할 경우 암종을 의심해야 한다. 암종은 세포의 크기, 핵소체의 뚜렷함, 세포질의 양 등에 따라 대세포암종과 소세포암종으로 나눈다. 아무리 신경내분비종양에서와 같은 배열양상이 유지되어 있고 염색질의 양상이 신경내분비종양에 가까운 경우라도 세포분열수가 많으면 신경내분비암종으로 분류하여야 한다.
기능성 여부에 따른 병리소견의 차이
기능성 췌장신경내분비 종양은 생물학적으로 활성된 펩티드 성분을 분비하며 인슐린, 가스트린, 글루카곤, 소마토스타틴, vasoactive intestinal polypeptide (VIP) 등이 이에 속하고, 비기능성 종양은 활성되어 있는 것은 아니지만 뉴로텐신(neurotensin) 또는 chromogranin A 등을 분비한다[5].
호르몬 기능발현과 연관된 호발부위는 가스트린종은 두부에서 VIP종은 미부에서 잘 생긴다. 크기는 인슐린종은 대개 2 cm 미만으로 작은 데 반해 나머지 호르몬 생성 종양은 크다. 그러나 종양의 크기와 호르몬으로 인한 증상의 초래 정도와는 관련이 없다. 비기능성 종양은 일반적으로 2 cm 보다 크며 흔히 5 cm 이상의 큰 종괴로 나타난다. 미소신경내분비종양인 경우 글루카곤종이나 췌장폴리펩티드(pancreatic polypeptide, PP) 종일 경우가 많다. 그러나 형태만으로는 종양의 기능성이나 생성 호르몬 유형 등을 예측할 수 없으며 각각의 호르몬 표지자에 대한 면역염색을 시행하여야 정확하게 알 수 있다. 다만 아밀로이드 형성은 인슐린종에서 사종체는 소마토스타틴종에서 나타나므로 분비호르몬 유형을 짐작할 수 있다.
악성 기준
내분비종양의 하나이므로 세포의 비정형이나 다형성 여부는 중요하지 않으며 이와 관계없이 전이가 있을 경우 악성이라고 할 수 있다. 전이는 간이 가장 흔하고 림프절로도 전이한다. 전이가 없더라도 육안적으로 장기 주변의 지방조직 내로 침윤하여 위성결절을 만들거나(Fig. 6A) 주변 십이지장 및 총담관, 비장 및 대혈관 등 주변장기로 직접 침윤할 경우(Fig. 6B) 악성이라고 진단할 수 있다. Ki67 증식지수가 악성능과 직접 상관관계를 가지며[1] cytokeratin 19가 침습성의 표지자로 사용될 수 있다[9]. 기타 종양의 크기가 2 cm 이상이거나 거짓피막 내 또는 주변에서 신경주위침윤이나 혈관침윤 등이 나타나면 의미가 있다. 그밖에 괴사, 프로게스테론 수용체 소실, 비배수체, 대립유전자 소실, CD44 과발현, p27발현 등이 있으나 그 의의는 낮다. 분자유전변화 중에서 DNA 복제수가 인슐린종에서는 가장 민감하고 임상경과를 예측할 수 있는 효과적인 표지자로 알려져 있다[10]. 또한 현미부수체불안정성이 종양 발생 초기 단계부터 관여하며 신경내분비종양의 침습성과 성염색체의 소실과 상관관계가 있다고 밝혀졌다[11].
면역조직화학염색
신경내분비종양 표지자인 chromogranin A 및 synaptophysin에 양성으로 염색되며(Fig. 7) 신경내분비종양이라고 진단을 하기위해서는 최소한 이 중 한 가지 표지자에서 양성이어야 한다. 이외 CD56 (NCAM1)에도 대부분 양성이며, neuron specific enolase (NSE)는 특이도가 낮아서 유용성이 떨어진다. Chromogranin A는 혈중수치도 임상적으로 의미가 있어 종양의 약 60~80%에서 상승되어 있고 종양크기나 범위와 비례하므로 진단 추적관찰 및 치료반응을 알아보기 위해서도 사용될 수 있다[12,13]. Cytokeratin 유형에 따른 면역반응여부는 cytokeratin 8 및 18은 양성인 반면 cytokeratin 7 및 20은 음성이다. 베타카테닌(β-catenin)은 고형거짓유두종양에서는 핵염색을 보이는데 반해 신경내분비종양에서는 세포막염색을 보인다[14].
증상에 따라 해당 펩티드 호르몬 각각에 대한 염색에서 양성으로 염색된다. 그러나 면역염색에서 펩티드 호르몬이 염색되었다고 하여 해당 호르몬의 증상이 임상적으로 나타나는 것은 아니며 신경내분비종양의 기능성, 비기능성 분류는 임상증상의 유무에 따른 것이지 면역염색표지자에 따른 분류가 아니다. 즉 종양세포가 호르몬을 생성하더라도 생성되는 호르몬의 양이 극히 적어서 증상을 유발할 수준이 아니거나, 생성된 호르몬이 비활성 상태인 경우는 임상적으로 호르몬으로 인한 증상을 유발하지 않기 때문이다.
감별진단
췌장에서 발생할 수 있는 일차 종양이 모두 해당되나 특히 고형성거짓유두종양(solid-pseudopapillary neoplasm), 소엽세포암종, 췌장모세포종, 원시신경외분비종양, 관샘암종 등이 있으나 특징적인 종양세포의 형태 및 배열양상, 신경내분비 표지자 양성 소견 등으로 대부분 쉽게 감별이 가능하다.
때로 샘암종의 일부 세포가 샘상피세포의 특성과 신경내분비 특성을 동시에 갖고 있을 때 혼합 샘신경내분비암종이라고 할 수 있는데 샘암종 내 신경내분비세포가 흩어져 소수 발견되는 일은 드물지 않으므로 최소한 어느 한 가지 성분이 30% 이상을 차지할 때 혼합 샘신경내분비암종이라고 진단할 수 있다. 췌장의 경우는 소엽신경내분비암종(acinarneuroendocrine carcinoma), 관신경내분비암종(ductal-neuroendocrine carcinoma), 혼합 소엽-신경내분비-관암종(mixed acinar-neuroendocrine-ductal carcinoma) 등이 있을 수 있다.
병리보고서 작성
병리보고서에는 다음 네 가지를 꼭 언급해야한다. 첫째 종양의 조직분류 즉, 신경내분비종양인지 신경내분비암종 인지 여부를 쓰고, 둘째 종양의 등급(G1, G2, G3), 셋째 TNM 병기를 쓰고, 넷째 세포유형이나 호르몬 기능여부에 따른 활성도 즉 기능성 여부를 포함한다. 그 외 chromogranin A 및 synaptophysin과 같은 신경내분비 표지자에 대한 염색결과, 혈관 및 신경침윤 유무, 종괴의 완전 절제 유무, 림프절 수 및 전이여부 등을 언급한다. 주변 조직에서의 신경내분비세포의 증식, 즉 동일호르몬분비세포과다증식 등이 있다면 함께 기술한다.
전구병변 및 유전
췌장의 신경내분비종양은 대부분 산발적으로 발생하나 일부 유전증후군의 일부로서도 나타난다. 가장 흔한 것이 다발성 내분비 종양 제 1형(multiple endocrine neoplasia type 1, MEN-1)이나[1] 드물게는 von Hippel-Lindau (VHL) 증후군, 신경섬유종증 제 1형(neurofibromatosis type 1, NF1), 결절성 경화증 복합증후군(tuberous sclerosis complex, TSC) 등에서도 같이 나타날 수 있다[15].
MEN1 환자의 췌장은 약 5 mm 직경 이내의 작은 여러 개의 내분비종양이 나타나는데 이를 가리켜 미소샘종증(microadenomatosis)라고 부른다. 이러한 환자의 미소종양의 발생이전에 나타나는 분자병리변화를 조사함으로써 신경내분비종양의 발생에 관여하는 유전변화를 밝히려는 연구가 소수 있다. Vortmeyer 등[16]은 내분비샘이 아닌 췌관과 관련된 병변에서 11q13 LOH가 나타난 점을 확인하고 섬세포종양(islet cell tumor)의 전구병변은 섬(islet)이 아닌 췌관에서 비롯된다고 주장하였다. Klöppel 등[17]은 모든 미소샘종 및 단일호르몬 내분비세포군집(monohormonal endocrine cell cluster)에서 MEN1 대립유전자 중 하나가 소실되었음을 발견하고 섬보다는 췌관에서 더 흔히 발생한다고 하였다. 그러나 아직까지 산발신경내분비종양에 대해서는 전구병변이 밝혀진 바가 없다.
병기 및 예후
American Joint Committee on Cancer (AJCC)에서 2010년 새로 발간한 TNM 병기체계제 7판에 따르면 소화기 신경내분비종양을 샘암종과 구분하여 고유의 신경내분비종양 병기체계를 적용하였다. 그러나 소화기계 장기 중 췌장과 충수돌기는 제외되었다. 췌장의 신경내분비종양은 크기(2 cm 기준)와 주변장기로의 침윤정도에 따른 통상의 췌장 관암종 병기체제와 동일하게 적용하였다[18].
신경내분비종양은 굳이 암종이 아니더라도 악성 잠재능을 갖고 있는 종양이다. 약 1 cm 미만의 인슐린종은 대부분 단순 종괴절제술만으로도 양호한 임상 결과를 밟지만 2.4~17.9%에서 악성 행동양식을 따른다. 인슐린 외 다른 호르몬유형의 기능성 신경내분비종양이나 비기능성 신경내분비종양은 대체적으로 침윤성을 갖는다.
5년생존율은 양성 인슐린종일 경우는 약 97%까지 이르지만 췌장의 비기능성 전이 신경내분비암종은 30%로 낮다. 또한 최근 자료에 따르면 저분화 신경내분비암종의 경우 위장관의 샘암종과 그 예후가 비슷하다[19].




 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






