


INTRODUCTION
Microscopic polyangiitis (MPA) is an idiopathic autoimmune disease. It is characterized by systemic vasculitis that affects the small blood vessels, such as arterioles, venules, and capillaries, and it is associated with anti-neutrophil cytoplasmic autoantibodies(ANCA). MPA is one of the major causes of pulmonary-renal syndrome, along with Goodpasture’s syndrome, systemic lupus erythematosus, and Wegener’s granulomatosis. Depending on the extent of systemic vascular involvement, clinical findings can be rather variable with cutaneous, musculoskeletal, neurological and gastrointestinal symptoms, as well as renal and pulmonary ymptoms. Pulmonary involvement is seen in 25-55% of MPA patients, with hemoptysis being the predominant symptom as a result of alveolar hemorrhage. However, pulmonary involvement also consists of pulmonary infiltrates, pleural effusion, pulmonary edema, pleuritis, and interstitial fibrosis. Interstitial lung disease(ILD) is a less recognized pulmonary manifestation of MPA. It can present anywhere from years prior to years after MPA diagnosis. Glucocorticoid, cyclophosphamide and mycophenolic mofetil (MMF) are typically used for induction therapy, while azathioprine is used for maintenance therapy. We report a case of MPA associated with ILD in which the patient experienced different organ responses to immunosuppressive therapies.
CASE REPORT
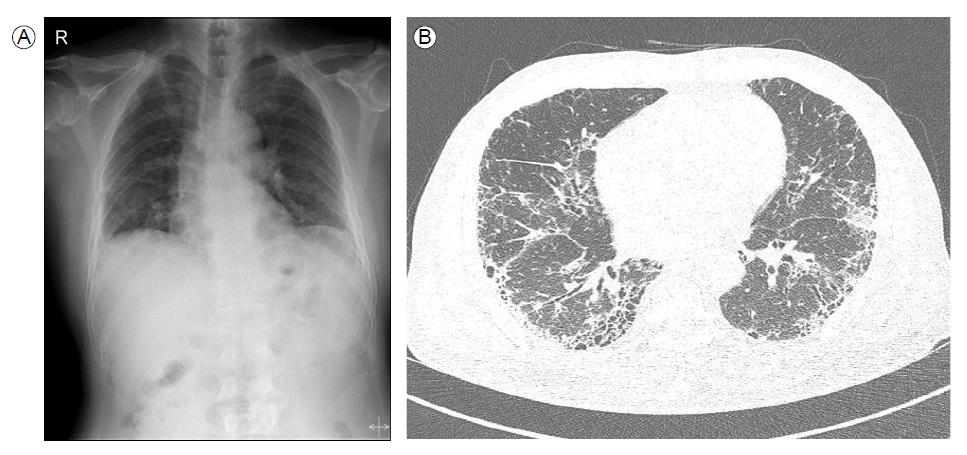
A 61-year-old man presented with a 2-month history of persistent cough with productive sputum and a 10-day history of dyspnea. He was an ex-smoker who stopped smoking 18 years ago. He had a history of hypertension and worked as a carpenter. On admission, auscultation of the lungs revealed bibasilar crackles without wheezing. Additionally, there was no clubbing, evidence of congestive heart failure, or cor pulmonale. A plain chest radiograph demonstrated reticulation in both lower lung fields (Fig. 1A), and computed tomography (CT) scan showed bibasilar reticulation and honeycombing in a peripheral distribution (Fig. 1B). Pulmonary function tests revealed a normal pattern with a mildly decreased diffusing capacity of the lung for carbon monoxide (DLCO). Blood tests revealed a white cell count of 20,640/mm3 (93.8% neutrophils), hemoglobin level of 10.5 g/dL, hematocrit level of 30.3%, and platelet count of 314,000/mm3. The serum creatinine level was elevated to 6.42 mg/dL, and the estimated glomerular filtration rate was 9.44 mL/min. Biochemical tests revealed a urea nitrogen level of 77 mg/dL, glucose level of 281 mg/dL, total protein level of 7.5 g/dL, albumin level of 2.9 g/dL, aspartate transaminase (AST) level of 25 IU/L, alanine aminotransferase (ALT) level of 37 IU/L, alkaline phosphatase level of 183 IU/L, and total bilirubin level of 0.59 mg/dL. The C-reactive protein level was 21.68 mg/dL. The 24-hr urine protein was 478.2 mg/day, and microscopic urinalysis showed several RBCs per high-power field. Furthermore, serologic investigation revealed perinuclear ANCA (p-ANCA) with a titer of 1:640. Anti-glomerular basement membrane (anti-GBM) antibodies and antinuclear antibodies (ANA) were negative.
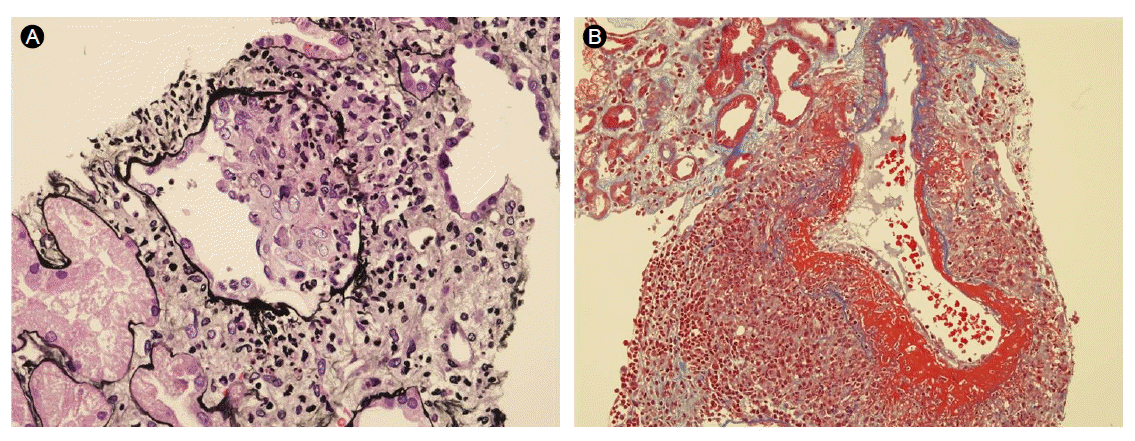
Renal biopsy revealed two global and four focal segmental sclerotic glomeruli out of 15 total glomeruli. Two cellular and fibrocellular crescent formations were present (Fig. 2A). Acute necrotizing arteritis was evident in the smaller vessels, showing mural and perivascular fibrinoid necrosis, with predominating neutrophils as well as occasional mononuclear leukocytes, including lymphocytes and macrophages (Fig. 2B). Immunofluorescence analysis showed no immune deposits.
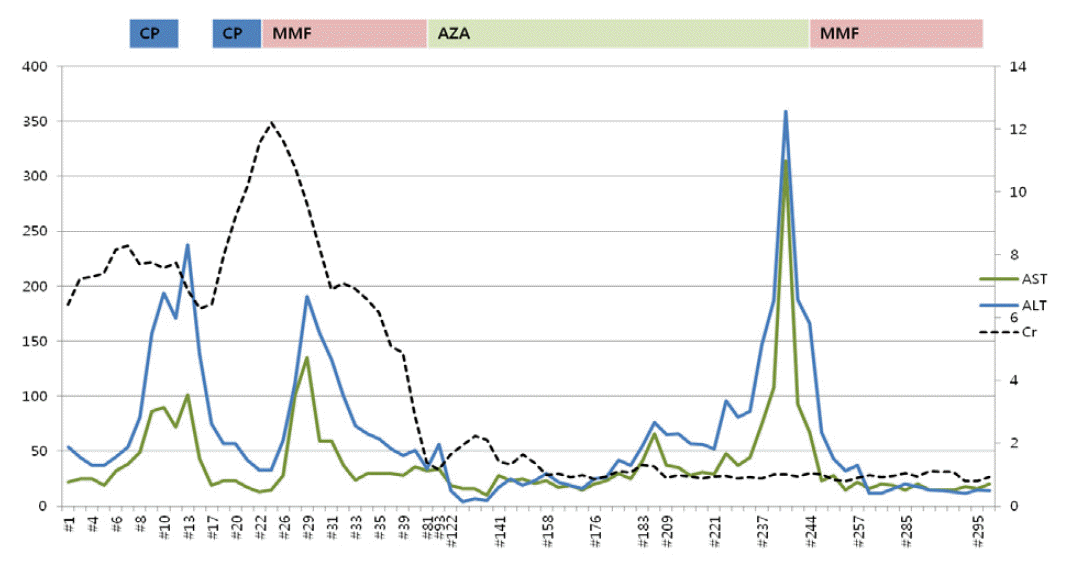
MPA was diagnosed on the basis of ANCA-positive serology and biopsy. Therefore, intravenous (IV) corticosteroids (methylprednisolone, 1 g IV for 1 day and 500 mg IV for the next 2 days) and oral cyclophosphamide (1 mg/kg/day, 50 mg/day) were initiated. Once intravenous steroid administration was completed, the patient was administered oral prednisone at a dose of 1 mg/kg/day daily. Additionally, the dose of cyclophosphamide was halved due to his impaired renal function and old age. Four days after starting cyclophosphamide, the patient’s liver enzyme levels were elevated, for which cyclophosphamide was considered to be responsible and was thus switched to MMF (1,500 mg/day). Following the switch to MMF, his liver enzyme levels settled and creatinine and urea nitrogen levels started to decrease (Fig. 3). He recovered sufficiently to be discharged and was followed up as an outpatient. After 3 months, MMF was switched to azathioprine as maintenance therapy.
Shortly after the switch to azathioprine, the patient was readmitted with aggravating dyspnea. Chest CT revealed worsening of pulmonary fibrosis with no cause of infection found. He continued immunosuppressive treatment, antibiotic therapy and supportive care, and his creatinine level gradually returned to the normal range, upon which blood testing for ANCA was negative. Approximately 70 days after initiating azathioprine, his liver enzyme levels, which had responded to the termination of azathioprine, began to increase.
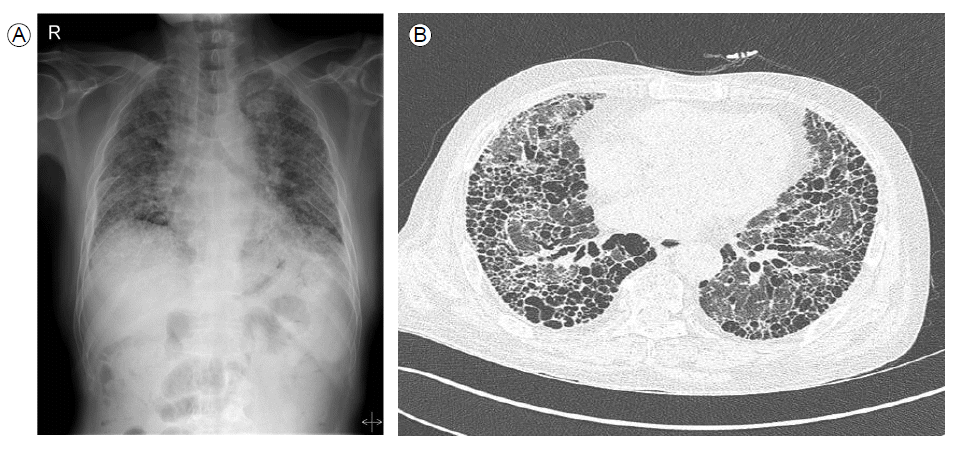
Although renal involvement was resolved, his pulmonary lesion worsened over time. Chest radiographs demonstrated dramatic progression of pulmonary recticulation and severe hypoxemia, and respiratory distress developed over 9 months. Despite being transferred to the intensive care unit and treated with aggressive supportive care and antibiotic therapy, he ultimately expired from hypoxic respiratory failure (Fig. 4).
DISCUSSION
The association between ILD and MPA has been reported recently. Although some reported cases of MPA were diagnosed concurrently with ILD [1], most MPA cases developed many years after diagnosis of ILD [2-4].
There are inconsistent results regarding mortality of MPA patients with ILD. Arulkumaran et al. [2,3] showed no differences in survival between MPA patients with ILD and those without ILD. On the other hand, some reports showed a significantly greater all-cause mortality among MPA patients with ILD than those without ILD [5,6].
In our case, ILD and MPA were diagnosed concurrently and treated with immunosuppressive agents, including steroid pulse therapy, cytotoxic drugs, and antimetabolites. We achieved a complete renal response but no response to ILD. We used a half-dose of cyclophosphamide due to the patient’s impaired renal function and old age, but MMF was substituted because of hepatotoxicity. Azathioprine was used for maintenance therapy but, after 3 months, it also showed hepatotoxicity and was switched back to MMF [7,8]. The MMF was dosed according to the recommendations of the European League Against Rheumatism, and the fact that we had a complete renal response showed that the adequate dose was delivered.
There is a lack of data for adequate dosing and regimens of immunosuppressants in MPA with ILD. Previous case reports used combined corticosteroid and cyclophosphamide as an induction therapy, while corticosteroid, azathioprine or MMF have been used as maintenance therapies for patients with MPA and ILD [1,2,4]. This regimen did not differ from our case.
We had a remarkable finding in that our patient was p-ANCApositive initially but then became seronegative according to the complete response of the kidney. It was the opposite of previous reports showing that ANCA was consistently positive, and irrelevant to the course of vasculitis, and played a direct role in the pathogenesis of ILD [1,9]. Taken together, ANCA might play an important role in the development of ILD, but it is not appropriate for monitoring disease activity. It also suggests the need for other markers, such as pulmonary function tests or high resolution CT (HRCT) scores [1,3,10].
According to previous studies [2-4,6], the causes of death varied even though the organs involved and the treatment regimens, such as aggravation of vasculitis, respiratory failure, infection and malignancy, were the same.
From this case study, we need to consider that the treatment response could be different for each organ, and lung involvement might be a crucial factor in mortality when treating patients with MPA and ILD. To improve the outcome of these patients, new treatment strategies for ILD and further randomized prospective controlled studies with larger patient groups are needed.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






