


서 론
폐동맥 고혈압은 유전성(heritable), 특발성(idiopathic), 또는 약물이나 기타 질환과 연관되어 발생되며, 연간 100만 명당 약 2명 정도의 유병률을 보이는 드문 질환이다[1]. 유전성 폐동맥 고혈압은 2008년 4th World Symposium on Pulmonary Hypertension (Dana Point, 2008)에서 개정된 폐 고혈압 분류체계에 따른 폐동맥 고혈압의 한 아형(Group 1.2)에 속한다.
유전성 폐동맥 고혈압의 경우, 약 70%에서 제2형 골 조형단백 수용체(bone morphogenetic protein receptor type 2, BMPR2)의 생식세포 돌연변이(germline mutation)가 발견된다[2]. 이보다는 드물지만 유전성 출혈성 모세혈관 확장증(hereditary hemorrhagic telangiectasia)의 원인 유전자이기도 한 activin recptor-like kinase type 1 (ALK1), endoglin (ENG)의 돌연변이가 발견되기도 한다[3]. 유전성 폐동맥 고혈압을 유발하는 것으로 알려진 이러한 유전자 돌연변이는 불완전 침투도를 보이는 상염색체 우성 유전병이기에 폐동맥 고혈압으로 진행 위험도는 평생에 걸쳐 10-20% 정도로 보고되고 있다[4].
증 례
증례 1
환 자: 이〇〇, 남자 32세
주 소: 운동 시 호흡곤란, 객혈
과거력: 없음
가족력: 아버지 52세 간암으로 사망
현병력: 32세 남자가 5년 전부터 악화되는 운동 시 호흡곤란(NYHA class II-III)과 간헐적인 객혈로 입원하였다.
검사 소견: 흉부 X-선상 경도의 심비대, 중심 폐동맥 확장 소견보였고, 심전도상 정상 동성리듬 91회/분, 우측 편위, 우심실 비대소견 보였으며, 폐기능 검사상 정상 범주에 속하였다. 심장 초음파상 우심방 및 우심실 확장 및 비대, 우심실 추정 수축기 혈압 84 mmHg, D-형 좌심실의 양상, 박출 계수 73%, 경도 폐동맥판막 부전이 관찰되었으며, 심낭 삼출은 없었고, Tricuspid Annular Plane Systolic Excursion (TAPSE) 1.57 cm, Tei index 0.86이어서 중증 폐 고혈압에 합당한 소견을 보였다. 이후 우심도자술을 시행하였으며, 평균 폐동맥압 81 mmHg, 폐동맥 쐐기압 14 mmHg, 심박출량 3.4 L/min, 폐혈관저항 1,576 dynesㆍsecㆍcm-5 소견을 보여 폐동맥 고혈압 진단하였다. 뒤이어 시행한 6분 보행검사상 보행거리는 462m이었다. 혈액 검사상 N-terminal probrain natriuretic peptide (NT-proBNP) 330.10 pg/mL, arterial blood gas analysis (ABG, room air)상 PCO2 36.7 mmHg, PO2 87.5 mmHg, SO2 97%였고, 심장 자기공명 영상 검사 및 흉부 전산화 단층 검사상 폐색전의 증거는 없었고, 폐동맥 확장소견을 보였다.
치료 및 경과: 환자 warfarin sodium, bosentan hydrate 지속복용하면서 외래 추적관찰 중이다.
증례 2
환 자: 이〇〇, 여자 29세
주 소: 운동 시 호흡곤란, 전신 부종
과거력: 없음
가족력: 내원 12개월 전 오빠가 폐동맥 고혈압 진단받음.
현병력: 29세 여자가 2011년 3월 자연분만 후에 발생하였으며 이후 악화된 전신부종과 운동 시 호흡곤란(NYHA class III)을 주소로 내원하였다.
검사 소견: 흉부 X-선상 심비대 및 중심 폐동맥 확장 소견보였고, 심전도상 정상 동성리듬 87회/분, 우측 편위, V2-6상 T파 역전 소견 보였으며, 폐기능 검사상 비교적 정상범주에 속하였다. 심장 초음파상 우심방 및 우심실 확장 및 비대, 우심실 추정 수축기 혈압 125 mmHg, 박출 계수 71%, D-형 좌심실의 양상을 보였고, 중등도 삼첨판 폐쇄부전 및 중등도의 심낭 삼출이 관찰되었으며, TAPSE 1.4 cm, Tei index 0.60이어서 중증 폐 고혈압에 합당한 소견을 보였다. 이후 시행한 우심도자술상, 평균 폐동맥압 83 mmHg, 폐동맥쐐기압 6 mmHg, 심박출량 3.9 L/min, 폐혈관저항 1,579 dynesㆍsecㆍcm-5 소견 보여 폐동맥 고혈압 진단하였다. 뒤이어 시행한 6분 보행검사상 보행거리는 225 m였다. 혈액검사상 NT- proBNP 1916.00 pg/mL, ABG (room air)상 PO2 63.4 mmHg, PCO2 29.4 mmHg, SO2 92%였고, 심장 자기공명영상 검사 및 흉부 전산화 단층 검사상 폐색전의 증거는 없었고, 폐동맥 확장소견보였다.
치료 및 경과: 환자 furosemide, spironolactone, bosentan hydrate, digoxin, warfarin sodium 복용하며 증상이 호전되어 외래 추적관찰 중이다.
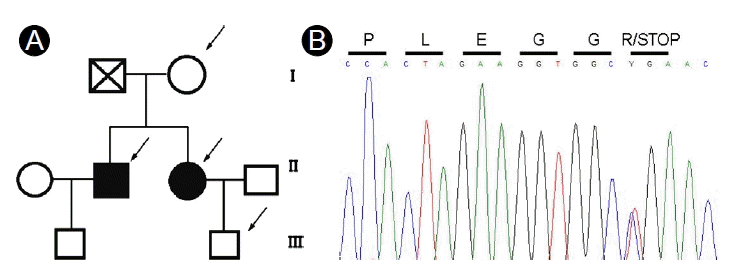
유전성 폐동맥 고혈압에 관하여 환자의 동의 및 미성년자의 경우 보호자 동의하에 전혈(whole blood)을 이용한 유전자 검사를 시행하였으며, PCR direct sequencing을 이용하여 BMPR2 유전자의 exon과 exon주변 intron의 염기서열에 대해 조사하였다(Fig. 1).
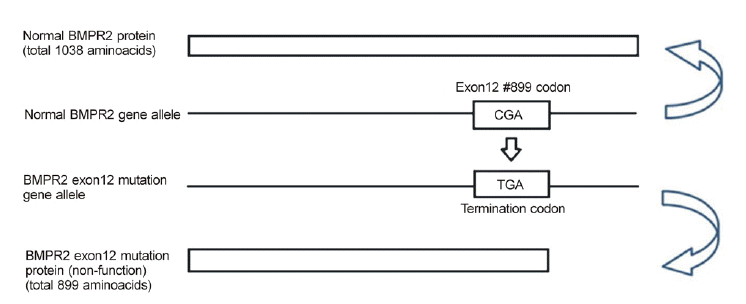
폐동맥 고혈압이 있는 남매 모두에서 두 개의 BMPR2 대립 유전자(allele) 중 하나의 대립 유전자에서 동일한 돌연변이가 발견되었다(c.2695C > T, p.R899X). 원래 BMPR2 단백질은 1038 아미노산으로 이루어져 있으나, 이 남매의 BMPR2유전자의 exon12에서 발견된 돌연변이는 899번째 아미노산 Arginine을 coding 하는 CGA codon이 TGA (termination codon)으로 바뀌면서 899번째 아미노산에서 조기 종결되어 정상 기능을 할 수 없는 단백질이 만들어질 것으로 추정된다(Fig. 2). 또한 이러한 BMPR2 유전자 돌연변이가 남매의 50세 어머니에게서 발견되었으나, 폐 고혈압의 증상 및 임상적, 검사상의 증거는 발견되지 않았다. 남매의 아버지는 52세 간암으로 사망하였으나, 평소 폐 고혈압을 의심할 만한 임상증상은 호소하지 않았다고 한다. 남매 각각의 자녀에 대해서 시행한 유전자 돌연변이 검사상에서 남환의 8개월된 아들에게서는 BMPR2 유전자 돌연변이는 발견되지 않았으나, 여자 환자의 5개월된 아들에게서는 BMPR2 유전자 돌연변이가 확인되었다.
고 찰
폐 고혈압(pulmonary hypertension)은 심박출량이 정상 혹은 저하되지 않은 상태에서 안정 시 침습적으로 측정한 평균 폐동맥압 > 25 mmHg으로 정의한다[5]. 2008년 4th World Symposium on Pulmonary Hypertension (Dana Point, 2008)에서 개정된 폐 고혈압 분류 체계에 따르면, 좌심장 질환에 의한 폐고혈압(group 2), 폐질환/저산소증에 의한 폐 고혈압(group 3), 만성 폐혈색전 폐 고혈압(group 4), 불명확한 다 요인적 기전에 의한 폐 고혈압(group 5)으로 구분하며, 폐동맥 고혈압은 group 1으로 분류하였다. 폐동맥 고혈압(pulmonary arterial hypertension)은 폐동맥계에서 기인한 폐 고혈압의 아형을 말하며, 따라서 혈역학적으로 정상 폐동맥 쐐기압, 즉 ≤ 15 mmHg 기준이 추가된다[5]. 유전성(heritable) 폐동맥 고혈압은 group 1.2로 분류하고, 이중 돌연변이와 연관된 유전자에 따라 BMPR2 (1.2.1), ALK1, endoglin (1.2.2), unknown (1.2.3)으로 분류되며, 여기서 주목할 점은 4차 폐 고혈압 분류에서 기존 3차 폐 고혈압 분류체계의 가족성(familial) 폐동맥 고혈압을 유전성(heritable) 폐동맥 고혈압으로 변경하였다는 점이다. 이는 가족력이 없는 특발성 폐동맥 고혈압 환자에게서 약 11-40% 정도 BMPR2 돌연변이가 발견되었고[7], 가족성 폐동맥 고혈압 중 약 30%에서 BMPR2 돌연변이가 발견되지 않음에 따라, 새 분류 체계에서는 가족성 폐동맥 고혈압이란 용어가 사라지게 되었다. 따라서 유전성 폐동맥 고혈압은 생식세포 돌연변이(주로 BMPR2 돌연변이 및 그외 ALK1이나 endoglin 돌연변이 등)를 지닌 특발성 폐동맥 고혈압과 생식세포 돌연변이 유무에 관계없는 가족력이 있는 경우로 정의한다[3].
유전성 폐동맥 고혈압은 일반적으로 최소 1.7:1의 비율로 여성에서 더 빈번히 나타나며[1], 유전성 폐동맥 고혈압은 평생에 걸쳐 10-20% 정도 발병 위험도를 지닌, 불완전 침투도를 가진 상염색체 우성 유전형질로 유전된다[4]. 이러한 불완전 침투도의 원인은 명확하지는 않으나, 호르몬을 포함한 환경적 요인, 유전자간의 상호작용, 질환에 대한 취약도 등이 거론되고 있다[8]. 본 증례의 남매 환자의 경우, BMPR2 exon12 돌연변이를 지닌 보인자(Carrier)이나 폐동맥 고혈압을 보이지는 않는 어머니로부터 BMPR2 exon12 돌연변이가 유전된 것으로 보여지며, 이어서 여자 환자의 아들로 유전된 것으로 판단된다. 하지만 어떠한 요인으로 이 유전자 돌연변이가 어머니에게서 폐동맥 고혈압으로 발병하지 않고, 남매에게서 폐동맥 고혈압으로 발병하게 되었는지는 명확하지 않다. 본 증례 환자들에 앞서, 2006년 국내에서 3차 폐 고혈압 분류체계에 따른 가족성(familial) 폐동맥 고혈압 환자로 의심되는 어머니와 아들의 증례를 보고한 적이 있다[6]. 해당 증례는 당시 1994년 폐동맥 고혈압으로 진단받은 어머니가 1997년 사망한 뒤, 6년 후 2003년 아들이 폐동맥 고혈압으로 진단받고 치료, 추적관찰 중 2004년 사망하여 가족성 폐동맥 고혈압 환자로 보고하였으며, 따라서 이 환자들을 대상으로 유전자 검사를 진행하지는 못하였다.
유전성 폐동맥 고혈압을 확인하는 것은 다음과 같은 임상적 의의를 갖는다. 첫 번째, 유전성 폐동맥 고혈압과 특발성 폐동맥 고혈압은 유사한 임상경과를 보이며, 생존율도 비슷하나, 유전성 폐동맥 고혈압이 상대적으로 더 어린 나이에 발병하며, 진단 당시 더 심각한 혈역학적 장애를 보인다는 점이다[9]. 이는 최근 연구에 따르면, BMPR2 유전자 돌연변이를 지닌 폐동맥 고혈압 환자들은 우심도자술 시 혈관확장제에 반응이 덜하며, 칼슘채널 길항제의 치료 효과 또한 덜효과적인 것으로 나타났다[9]. 두 번째, 폐동맥 고혈압의 가족력이 있는 가계에서 후세대로 갈수록 발병연령 및 사망연령이 낮아지는 경향을 보이는 것으로 알려져 있다[10]. 따라서 유전성 폐동맥 고혈압 환자의 가족 내 다른 구성원에게서 폐동맥 고혈압의 발병 가능성을 설명하고, 이를 바탕으로 정확한 유전적 정보에 근거하여 가족계획과 연관된 정보를 제공하는 것이 필요하다는 점이다. 증례의 여환의 아들의 경우, BMPR2 유전자 돌연변이를 지니고 있고 이로 인해 평생에 걸쳐 10-20% 정도의 확률로 폐동맥 고혈압으로의 발현가능성이 있으며 가족력이 있으므로, 향후 지속적인 추적관찰이 필요하다. 따라서 가족력이 있는 폐동맥 고혈압 환자나 특발성 폐동맥 고혈압 환자의 경우, 단 유전자 검사에 대한 윤리적, 제도적 보완이 이루어진다는 전제하에 의료진은 이들에게 유전자 검사의 필요성에 대한 내용을 설명할 필요가 있다.
 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Download Citation
Download Citation Print
Print






